
Abstract
The human gut microbiome plays a crucial role in human health and efforts need to be done for cultivation and characterisation of bacteria with potential health benefits. Here, we isolated a bacterium from a healthy Indian adult faeces and investigated its potential as probiotic. The cultured bacterial strain 17OM39 was identified as Enterococcus faecium by 16S rRNA gene sequencing. The strain 17OM39 exhibited tolerance to acidic pH, showed antimicrobial activity and displayed strong cell surface traits such as hydrophobicity and autoaggregation capacity. The strain was able to tolerate bile salts and showed bile salt hydrolytic (BSH) activity, exopolysaccharide production and adherence to human HT-29 cell line. Importantly, partial haemolytic activity was detected and the strain was susceptible to the human serum. Genomics investigation of strain 17OM39 revealed the presence of diverse genes encoding for proteolytic enzymes, stress response systems and the ability to produce essential amino acids, vitamins and antimicrobial compound Bacteriocin-A. No virulence factors and plasmids were found in this genome of the strain 17OM39. Collectively, these physiological and genomic features of 17OM39 confirm the potential of this strain as a candidate probiotic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bacteria in the colon have been correlated with healthy phenotype and some with diseased phenotypes (Rojo et al. 2016). Moving ahead from correlations, recent studies have demonstrated the beneficial effect of specific or consortia of microbes on human health (Atarashi et al. 2013; Subramanian et al. 2014; Plovier et al. 2016). Modulating the gut microbiota for improving and or maintaining a healthy status is a promising avenue and can be approached from various aspects like diet and specific probiotics or consortia of probiotics (Rojo et al. 2016; O’Toole et al. 2017).
For a long time, bacteria from genera like Lactobacillus, Lactococcus, Bifidobacterium, Leuconostoc, Bacillus, Streptococcus, Clostridium and Escherichia were being used as general probiotics. However, recent advances in high throughput sequencing have revealed population-specific features of the human gut microbiome (Rastall et al. 2005; Consortium 2013; Falcony et al. 2016). Hence, identifying specific populations or indigenous beneficial bacteria will be an important aspect of designing microbiome-based therapeutics. Initial studies on the Indian microbiome have indicated a notable difference from other populations over the world (Bhute et al. 2016). This is hypothesised to be a consequence of its diverse genetic composition, dietary habits, cultural, religious affiliations and vast geographic scattering (Shetty et al. 2013). The current probiotics marketed in India for Indians are mostly non-indigenous strains and their efficacy is debated (Raghuwanshi et al. 2015).
In the present study, we screened bacterial strains isolated from faecal samples of healthy Indian subjects for their probiotic potentials. More than 125 different strains from 18 different genera were isolated using 35 culture media and different growth conditions. Among these, the strain 17OM39 of Enterococcus faecium showed properties of a potential probiotic. This work utilises an integrated approach of in vitro experiments and genomic analysis to characterise the metabolic characters of strain 17OM39.
Materials and methods
Isolation and preservation
Three self-declared healthy volunteers were selected for this study. Approval from the Institutional Ethics Committee (IEC) and consent from the subjects was taken prior to collection of the sample. Faecal samples were collected in a pre-sterile container and was immediately transported to the lab at 4 °C and processed within 6 h. For isolation of faecal bacteria, 1 g faces were transferred to 9 ml of sterile saline (0.85% sodium chloride, Sigma) and mixed well. The serial dilutions were subsequently prepared in sterile saline. Appropriate dilutions of the samples were plated on MRS media (HiMedia, Mumbai, India). Plates were incubated at 37 °C for 24 to 48 h under aerobic condition (Mishra et al. 2015). A pure culture of the isolate was preserved in 20% (v/v) glycerol as frozen stocks at − 80 °C.
Identification
Genomic DNA was extracted by using QIAGEN Blood and Tissue Kit (QIAGEN, USA) as per manufacturer’s protocol. DNA quantification was done by Nanodrop ND1000 (Thermo Scientific, USA). The 16S rRNA gene was amplified by using eubacteria specific primers 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492R (5′-ACG GCT ACC TTG TTA CGA CTT-3′) as described in the earlier study (Patil et al. 2015). Amplified PCR products were purified using polyethylene glycol (PEG)–NaCl precipitation (Evans 1990). Sequencing of both strands was done on an ABI 3730xl DNA analyser using the Big Dye terminator kit (Applied Biosystems, Inc., Foster City, CA). The sequence obtained was assembled using DNASTARPro, version 10 (Patil et al. 2015) and taxonomic identity were checked using the EZ-Biocloud server (Kim et al. 2012). Microscopy by SEM was performed stated elsewhere (Golding et al. 2016).
Characterisation of probiotic properties
Antibiotic susceptibility testing was carried out by disc diffusion method using the Dodeca Universal I & II kit (Hi-media, India). The isolate was tested for nine exoenzymes by standard microbiological methods viz. phosphatase by Pikovskaya’s agar base (Shen et al. 2016), urease by urease agar base (Vuye and Pijck 1973), lipase by tributyrin agar base (Yusof et al. 1989), cellulase by CMC agar (Kasana et al. 2008), protease by skimmed milk agar (Savijoki et al. 2006), gelatinase by gelatin medium (Whaley et al. 1982), catalase by effervescence of 6% H2O2 (Whittenbury 1964), nitrate reductase by colour change method (Tiso and Schechter 2015) and amylase activity by starch iodine test (Carrasco et al. 2016). Carbohydrate utilisation test was performed according to manufacturer’s instruction using HiCarbohydrate™ Kit (Sigma, India) consisting of 34 carbohydrates. The presence of any plasmid was checked by QIAGEN Plasmid Mini Kit (QIAGEN, USA). Bile and acid tolerance assay (Chou and Weimer 1999; Muller et al. 2011; Hassanzadazar et al. 2012; Tokatl et al. 2015), autoaggregation assays was performed by briefly 107 cells/ml of probiotic cells were harvested washed with PBS (pH 7.2) and re-suspended in the same buffer. Consecutively, bacterial suspensions were incubated at 37 ± 2 °C and monitored at different time intervals (0, 1, 2, 3, 4, and 5 h). The percentage of autoaggregation was expressed as A% = (A0 − At)/A0 × 100 where A0 represents the absorbance (A600 nm) at 0 h and At represents the absorbance at different time intervals (Schmidt and Hensel 2004), cell surface hydrophobicity (Saran et al. 2012), adhesion to human HT-29 cell line (Gyles and Boerlin 2014; Mishra et al. 2015), bile salt hydrolytic (BSH) activity (Zanotti et al. 2015), hypocholesterolemic activity (Sridevi et al. 2009), resistance to hydrogen peroxide (Halliwell et al. 2000; Wu et al. 2014; Forman et al. 2016) exopolysaccharide production (Abdhul et al. 2014), haemolytic activity (Ike et al. 1987; Semedo et al. 2003a, b) and serum resistance (King et al. 2009) was carried out as stated. Percentage cell survival was determined by calculating the ratio of amount of cell survived at the end of experiment (colony count) to the number of cell seed initially (colony count).
Antimicrobial activity of the strain 17OM39
The inhibitory activity assay was performed against Listeria monocytogenes (ATCC 13932), Pseudomonas aeruginosa (ATCC 15442), Streptococcus pneumonia (ATCC 49619) and Escherichia coli (ATCC 25922) as described in previous study (Pieniz et al. 2014).
Statistical analysis
All the experiments were done in triplicates. Mean values and standard deviation were calculated from the data obtained from triplicate assays. These data were then compared using Duncan’s Multiple Range Test (SPSS Ver. 10.0).
Genome sequencing and assembly
Genomic DNA extraction was followed as stated in the manufacturer’s protocol (QIAamp genomic DNA kit, Germany). The high-quality DNA was sequenced using Illumina MiSeq platform using 2 × 300 paired-end libraries. Reference-based assembly of quality-filtered reads was done using MIRA assembler version 4.9.3 (Chevreux and Wetter 1999). The genome of E. faecium T110 (GenBank ID: CP006030.1) was used as a reference.
Bioinformatics analyses
The draft genome sequence was annotated with Rapid Annotation using Subsystem Technology (RAST) version 4.0 (Overbeek et al. 2014) and the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al. 2016). Protein coding genes and tRNA and rRNA genes from the genomes were predicted using Glimmer version 3.02 (Delcher et al. 1999), tRNA_scan-SE (Lowe and Eddy 1997), and RNAmmer (Lagesen et al. 2007), respectively. Protein coding genes were also analysed by COG database (Tatusov et al. 2000) and Pfam domains were predicted using NCBI Batch CD-Search Tool (Marchler-Bauer et al. 2011). Presence of CRISPR repeats was predicted using the CRISPRFinder tool (Grissa et al. 2008). The origin of replication was predicted using ORIFinder (Luo et al. 2014). The open reading frames were obtained using the ORF finder tool (https://www.ncbi.nlm.nih.gov/orffinder/). Prophage sequences were predicted and annotated using PHASTER (Arndt et al. 2016). Bacterial insertion elements (ISs) were identified by ISfinder (Siguier et al. 2006). Horizontal gene transfer was detected by genomic island tool: Islandviewer (Juhas et al. 2009). Gene clusters of any bioactive compounds were identified by antiSMASH: antibiotics and Secondary Metabolite Analysis SHell (Medema et al. 2011). PlasmidFinder was used to search for plasmids within the genome (Carattoli et al. 2014). Codon usage analysis was done using General Codon Usage Analysis: GCUA (McInerney 1998). Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to predict the metabolic pathways from the genome (Ogata et al. 1999). The in silico DDH was done using the Genome-to-Genome Distance Calculator (version 2.1) server at http://ggdc.dsmz.de/ of Leibinz Institute DSMZ-German Collection of Microorganisms and cell culture. The average nucleotide identity comparisons were done using the ANI Calculator available at the EZ-Biocloud server (Lee et al. 2016). In silico analysis of complete bacterial genomes for RLFP was done at http://insilico.ehu.es/restriction/ (Bikandi et al. 2004).
Accession number(s)
This whole genome shotgun project has been deposited at GenBank under the accession LWHF00000000. The version described in this paper is version LWHF00000000 GenBank accession number for the partial 16S rRNA nucleotide sequence is KY682304.
Results
Microscopy and identification
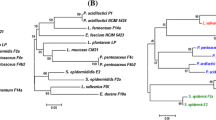
The bacterial cell morphology was examined using scanning electron microscope. The coccoid cells occur in pairs or short chains (Fig. S1). The coccoid cells of strain 17OM39 have a diameter of ~ 1.35 μm. The 16S rRNA gene of strain 17OM39 shows 99.92% similar to E. faecium type strain CGMCC (Fig. 1).

Phylogenetic relationship of strain 17OM39 with closely related taxa based on 16S rRNA gene sequences (16S rRNA gene sequence of Lactococcus lactis was used as out-group). The phylogenetic trees were constructed using the neighbour-joining method. The evolutionary distances were computed using the Kimura 2-parameter method and are in the units of the number of base substitutions per site. The rate variation among sites was modelled with a gamma distribution (shape parameter = 1)
Characterisation of probiotic properties
Exoenzymes, carbohydrate utilisation, antibiogram and plasmid determination
The strain 17OM39 was positive for protease, cellulase, catalase, amylase, lipase and nitrate reductase activities (Table 1). The strain was also able to metabolise most of the carbohydrate sources tested in this study few of them are lactose, xylose, maltose, fructose, dextrose, galactose, raffinose, trehalose, melibiose, sucrose, mannose and inulin (Table 2). The strain was sensitive to 19 of the 24 tested antibiotics and showed only intermediate resistance to 5 which were levofloxacin, ciprofloxacin, cefotaxime, gentamicin and furazolidone (Table 3). Absence of plasmids was confirmed by plasmid extraction method.
Bile, acid and hydrogen peroxide tolerance
Strain 17OM39 was able to grow in the bile concentration of 0.3% bile salt with 80% survivability after 24 h (Fig. 2a) and survive up to 1% (w/v) bile salt concentration (data not shown). The acid tolerance assay was carried out at pH 2.5 for 3 h depicting the condition and time taken by the food in the stomach with 74.02% survivability in these conditions (Fig. 2b). The strain was able to tolerate exposure to hydrogen peroxide for 6 h and tolerance declined gradually thereafter (Fig. 2e).

Results for the experiments conducted for characterisation of probiotic properties, where 17OM39 is Enterococcus faecium and control is Lactobacillus rhamnosus GG. a Bile tolerance test shows the survival of 17OM39 and control in 0.3% bile salt. b Acid tolerance test shows the survival of 17OM39 and control at pH 2.5 for 3 h. c Percentage autoaggregation capacity of 17OM39 and control. d Serum resistance test conducted for 17OM39 and control. e Peroxide resistance test conducted for 17OM39 and control in hydrogen peroxide. Also, error bar indicates a standard deviation (SD) and percentage cell survival was determined by calculating the ratio of amount of cell survived at the end of experiment (colony count) to the number of cell seed initially (colony count)
Adhesion ability and exopolysaccharide production
Autoaggregation of the strain 17OM39 was observed to be 32% (Fig. 2c, Table 4), while the cell surface hydrophobicity was observed with hydrocarbons such as hexane (32%), toluene (42%) and hexadecane (45%). Adhesion to human HT-29 cell line was found to be 57 and 60% for strain 17OM39 and control, respectively. In addition, microscopic observations revealed strong adhesion (Fig. 3). The strain 17OM39 is also capable of producing exopolysaccharide (Table 4).

Results for cell adhesion assay performed on Human HT-29 Cell line. The upper panel shows images taken under fluorescence microscope 60× oil immersion, while the lower panel represents the images taken under normal microscope. a, a1 Negative control without any bacterial cell added. b, b1 Strain 17OM39. c, c1 Positive control Lactobacillus rhamnosus GG
BSH activity and cholesterol removal
A 17-mm-diameter zone of clearance was observed demonstrating the ability of strain 17OM39 to hydrolyse bile. The ability to hydrolyse bile was confirmed by the ninhydrin method (Table 5). The BSH activity of strain 17OM39 was equal to that of the control strain Lactobacillus rhamnosus GG. It was observed that the active cells were able to remove cholesterol better than resting and dead cells (Table 5).
Pathogenicity testing
Pathogenicity testing included the investigation of haemolytic activity and serum resistance. The strain 17OM39 exhibited alpha haemolytic activity and was found susceptible to human serum with 0.01% survival after 3 h (Fig. 2d).
General genome features
More than 5 million good-quality paired-end reads were obtained, which accounted for an approximate 100× sequencing coverage. The complete genome of E. faecium 17OM39 consisted of 2,696,332 bp (2.69 Mb) with an average GC content of 38.5%. Genome consisted of 2865 genes and 2639 ORFs were identified. The general genome information is in Table S1 and the genome atlas in Fig. 4. Within the genome, we identified 78 RNA genes: 5S RNA (four copies), 16S RNA (six copies) and 23S RNA (two copies). The relative synonymous codon usage found in the genome is presented in Table S2. Protein coding genes were analysed by COG database on WebMGA and revealed 76.60% code for major cellular processes and signalling, information storage, processing and metabolism, while the remaining 23.4% genes were poorly characterised (Table S3). Strain 17OM39 showed 81% DNA–DNA relatedness, 97.83% ANI to the marketed probiotic E. faecium strain T110. The in silico genome RFLP with SnaI gave 28 and 31 bands for strain 17OM39 and T110, respectively.

Genome atlas of strain 17OM39. The atlas represents a circular view of the complete genome sequence of Enterococcus faecium 7OM39. The circle was created by using CG viewer. Innermost circle 1 shows GC-skew (black). Circle 2, 3 shows CDS’s (blue). Circle 4 represents genes for stress and Allied metabolism (pink). Circle 5 shows genes for carbohydrate and amino acid metabolism (green). Genes imparting probiotic properties are shown in circle 6 (Sky blue)
Mobilome
In silico analysis of the genome of E. faecium 17OM39 revealed two prophages; one complete and the other is an incomplete prophage. The complete phage region has genes encoding for lysin, tail, capsid, head, portal, terminase and integrase, while the incomplete phage has transposase, tail and lysine (Table 6 and Fig. 5a). Seven putative transposase genes, representing three different families namely IS4, IS256 and IS982 were identified in E. faecium 17OM39. These families appeared in duplicate copies and are highly conserved (Table 7).

In silico analysis performed. a Predicated prophages in the genome of Enterococcus faecium 17OM39 with position using PHASTER tool. b Predicted genomic islands using Island Viewer tool
Clustered regularly interspaced short palindromic repeats (CRISPR) elements were not found in the genome of strain 17OM39 genome. A total of 10 genomic island regions were identified and none of these regions was found to contain any antibiotic resistance gene (Fig. 5b). Our analysis suggested that these regions harboured genes mainly encoding few ribosomal, hypothetical and most of the viral proteins. However, some genomic islands contained genes encoding for death on curing protein, glutathione synthesis, methionine ABC transporter and gluconate dehydratase (EC 4.2.1.39). The regions in probable genomic islands are described in Table S4. Moreover, the genome was devoid of origin of transfer (oriT or bom) and transfer (tra) genes essential for plasmid maintenance.
Bacteriocin gene cluster
The genome of E. faecium 17OM39 harboured seven secondary metabolite gene clusters as observed using antibiotics and Secondary Metabolite Analysis Shell tool (antiSMASH). These clusters belong to microcin and bacteriocin family, while only one cluster was observed with a significant hit of 66% similarity to bacteriocin, located at nucleotide position 2312434-2313194 in the genome (Fig. S2). The bacteriocin has three domains viz. enteriocin induction factor (1-147), enteriocin A immunity protein (251-589) and enteriocin A (564-761). Based on the previous evidence that such type of bacteriocin exhibits activity against L. monocytogenes specifically, antimicrobial activity assay was performed against L. monocytogenes (ATCC 13932) along with P. aeruginosa (ATCC 15442), Streptococcus pneumonia (ATCC 49619) and E. coli (ATCC 25922). It was observed from the experiment; the antimicrobial activity was against L. monocytogenes (9.2 ± 0.2 mm as zone of clearance). In vitro experiment supports the presence activity of this extracellularly secreted bacteriocin as it inhibited the growth of L. monocytogenes. The bacteriocin is being further characterised in our lab.
Genome-based metabolic capabilities
We screened the genome sequence of the strain 17OM39 for metabolic capabilities to understand the cellular processes accounting for potential probiotic nature.
Amino acid synthesis
Analyses of genome report the potential of strain 17OM39 to synthesise amino acids such as cysteine, serine, and aspartate. From the three amino acids (cysteine, serine, and aspartate), seven other amino acids and derivatives could be generated (Kleerebezem et al. 2003). The presence of particular genes in a pathway was assessed and metabolic pathway for lysine was constructed as shown in Fig. S3. The genome harboured genes encoding for Beta-lyase (metC) and L-serine dehydratase (EC 4.3.1.17) to synthesise cysteine and serine from pyruvate. Detailed information about amino acid synthesis can be found in Supporting Text.
Allied metabolism
Sulphate permease (permits SO24− into the cell) and serine acetyltransferase (EC 2.3.1.30) the enzymes required for sulphate incorporation from H2S for the synthesis of L-serine were traced in the genome. An alternative pathway of ammonia assimilation involving glutamine synthetase (GS- EC 6.3.1.2) was found in the genome. It was observed that based on the genes present, thiamine synthesis can be formed by series of enzymes as shown in Fig. S4. The metabolic pathway for folate synthesis was traced into the genome from 7,8 dihydropteroate to folate by seven enzymes (Fig. S5). Detailed information about allied metabolism can be found in Supporting Text.
Proteolytic system
Three types of proteolytic systems were found in strain 17OM39, i.e. oligo peptidases, aminopeptidases and carboxypeptidases specific for D-alanine-D-alanine. The type M (metallo) peptidase group consisted of two members: PepO, PepF. Group type S (serine) consisted of only five members—glutamyl aminopeptidase (EC 3.4.11.7); methionine aminopeptidase (EC 3.4.11.18); aminopeptidase S (Leu, Val, Phe, Tyr preference - EC 3.4.11.24); aminopeptidase YpdF (MP-, MA-, MS-, AP-, NP- specific); aminopeptidase C (EC 3.4.22.40) and tripeptide aminopeptidase (EC 3.4.11.4). In addition to these, CAAX protease family was detected. These proteases share several conserved motifs and most members are likely membrane-bound endopeptidases with no common specificity. Detailed information about the proteolytic system can be found in Supporting Text.
Carbohydrate metabolism
The 17OM39 genome encodes a large variety of genes related to carbohydrate such as mono-, di- and oligosaccharides; an amino sugar; organic acids and sugar alcohols. Annotation from RAST and PGAP has shown the genes present in Entner-Doudoroff pathway, pentose phosphate pathway, glycolysis and gluconeogenesis and pyruvate metabolism I and II. The strain 17OM39 was able to utilise xylose, maltose, dextrose, raffinose, melibiose, arabinose, inulin, sodium gluconate, glycerol, inositol, sorbitol, mannitol, adonitol, rhamnose, esculin, D-arabinose, citrate, malonate and sorbose in the experiment conducted. Moreover, genomic analysis further confirms the presence of these genes within the genome. We could also trace for lactose, fructose, galactose, trehalose, sucrose, mannose, salicin, arabitol and cellobiose along with the transports, which was see in the API strip assay. Figure 6 shows carbohydrate utilisation found in the genome. Detailed information about carbohydrate metabolism can be found in Supporting Text.

Carbohydrate utilisation found in the genome. The diagram shows transporters and enzymes, as predicted by the putative genome annotation. For transporters, blue and red indicates a putative PTS transporter; orange a putative ATP-binding cassette (ABC) transporter; and pink a galactoside permease
Stress response
The genome of E. faecium 17OM39 encodes a number of stress-related proteins, including numerous proteases involved in the stress response and has highly conserved SOS regulon genes. Also, heat shock proteins were identified in the genome of 17OM39; these include the highly conserved class I heat shock genes responsible for maintaining the integrity of cellular proteins under stress conditions (GroES and GroES operons) and the conserved class III Clp proteases ClpB, C, E and X. The F1F0-ATPase system and superoxide dismutase (SOD) have been identified in this genome. The genome also harbours universal stress protein family, non-specific DNA-binding protein (Dps), iron-binding ferritin-like antioxidant protein and ferroxidase (EC 1.16.3.1), redox-sensitive transcriptional regulator and peroxide stress regulator to organic hydroperoxide resistance protein were annotated. Glutathione redox cycle along with glutathione reductase (EC 1.8.1.7), NADH peroxidase (npx; EC 1.11.1.1) and recA, involved in repairing DNA damage was also encoded by the genome. Cold shock protein (cspA (four copies), B, D) were observed in the genome. Lastly, a gene relA was identified that encodes an enzyme putatively involved in osmotolerance via synthesis and hydrolysis of (p)ppGpp.
Regulation systems
One major (rpoD) and one putative minor alternative sigma factor (sigV) was found in the genome sequence of strain 17OM39, essential for the bacterial cell to adjust its metabolism to changing environmental conditions. The housekeeping Rrf2 family transcriptional regulator, group III and DNA-directed RNA polymerase with its all subunit were observed in the genome. Different operon transcriptional repressors were also found in the genome such as trehalose-, maltose- (MalR), fructose-, phosphosugar-binding (RpiR family) and heat-inducible repressor (HrcA) and also repressors for sucrose (ScrR), ribose, galactose and lactose operon were found. The genome also has arginine pathway regulatory protein (ArgR), purine nucleotide synthesis repressor, arabinoside (GntR) and biotin operon repressor, osmoregulation-related genes, zinc (ZUR) and ferric (FUR) uptake regulation protein. Also, few repressors for DNA were detected: DNA-binding response regulator (OmpR family), DNA-binding heavy metal response regulator and DNA-binding response regulator AraC family.
Probiotic genes associated with the genome
The cholylglycine hydrolase (EC 3.5.1.24) gene responsible for bile salt hydrolysis action was identified in two copies within the genome. Fibrinogen and collagen-binding protein were found in the genome allowing them to bind the GI tract, suggesting an important role in adhesion and colonisation in intestinal mucosal surfaces. Aggregation-promoting factor (apf) is a protein that has been associated with self-aggregation and maintenance of cell shape and was identified in the strain 17OM39 genome. While the EPS cluster consisted of 14 genes including the highly conserved proteins EpsB–EpsF was located in the genome. Also, the resistance to hydrogen peroxide is imparted by genes alkyl hydroperoxide reductase (ahp) and NADH peroxidase (npr) were found in the genome, while this activity was also evident as shown by the experiments. In addition, based on the previous studies, we also screened for a set of genes involved in imparting important probiotic functions, as described in Table 8.
Absence of virulence determinants
We screened for the presence of virulence genes that are commonly known to be associated with other enterococci. Cytolysin (cyl), aggregation substance (as), gelatinase (extracellular metalloendopeptidase EC 3.4.24.30), hyaluronidase (efm), the two cell wall adhesins (efaAfs and efaAfm), two sex pheromones (cob and ccf), hemolysis (hlyA), serum resistance-associated gene (sra) and any other virulence factors could not be traced into the genome.
Discussion
As a part of our research to catalogue and characterise the bacterial populations within the Indian population, we isolated an indigenous strain of E. faecium and comprehensively tested for its probiotic potential. For a successful colonisation of bacteria, it needs to have the ability to tolerate low pH, presence of bile and tolerate oxidative stress. The human intestinal tract has both facultative and obligate anaerobic bacteria. Addition of facultative bacteria to synthetic microbial communities has been shown to improve colonisation resistance in mice (Brugiroux et al. 2016). Hence, it is important to investigate the commensal facultative anaerobes from the human intestinal tract. In the present study, we use a combination of in vitro and in silico approaches to test the indigenous bacterial strain 17OM39 for its probiotic attributes.
First, we tested the acid tolerance and found that strain 17OM39 was able to tolerate a low pH of 2.5 for 3 h. This is an important character for a bacterium to be able to survive and become part of the natural microbial community. In many cases, bacteria that are unable to tolerate low pH, bile and transition through the intestinal tract require microencapsulation to aid successful delivery (Brugiroux et al. 2016). Along with experimental evidence, we identified the genes known for encoding proteins involved in acid tolerance (lytr and ehr) within the genome (Lebeer et al. 2008). We also identified genes encoding for bile resistance proteins (cdpA, clpE and dps) (Lebeer et al. 2008) and genes responsible for bile hydrolysis (bsh) (Begley et al. 2006). In vitro assays demonstrate that our strain is able to tolerate low pH and has 80% survivability in vitro to 0.3% bile. This suggests that our strain may not need protective encapsulation for successful transition through the host intestinal tract.
Next, we tested the aggregation and adhesion properties of strain 17OM39 as it is an important feature for long-term colonisation in the host intestinal tract. Our strain showed only marginally low adhesion and autoaggregation than the control L. rhamnosus GG, a known probiotic (P value < 0.51, paired nonparametric “t test”). Genome analysis revealed the presence of gene encoding for aggregation-promoting factor. These proteins are associated with self-aggregation, maintenance of cell shape and enhance the gastrointestinal persistence of the organism in vivo (Jankovic et al. 2003; Goh and Klaenhammer 2010). The genome of strain 17OM39 consisted of genes encoding for fibrinogen and collagen-binding protein suggesting its ability to adhere to host cells. We confirmed this by adhesion assay using human HT-29 cells and analysis of cells surface showed a hydrophobic nature which is helpful for adhesion. The adhesion of the strain to the intestinal mucosa helps to increase the persistence of probiotic cells in the gut and therefore enabling colonisation and competitive exclusion of pathogens (Greene and Klaenhammer 1994; Ouwehand and Salminen 2003). Further, the presence of exopolysaccharides (EPS) gene cluster and in vitro results for production of EPS by strain 17OM39 confirms its ability to form EPS which could aid in higher tolerance to environmental stress in gastrointestinal tract (Mozzi et al. 2009; Ciszek-Lenda et al. 2011). Also, the resistance to hydrogen peroxide is imparted by genes alkyl hydroperoxide reductase (ahp) and NADH peroxidase (npr) were found in the genome, while this activity was also evident as shown by the experiments. In addition, based on the previous studies, we also screened for a set of genes involved in imparting important probiotic functions, as described in Table 6.
The genome analysis of strain 17OM39 for insights into its carbohydrate metabolism revealed a diverse array of gene encoding for utilising numerous carbon sources for energy and growth. Investigations for carbohydrate (CHO) metabolism has been carried out using a two-step integrated approach which includes the genomic data such as presence of genes encoding for important enzymes involved in CHO metabolism along with the genes encoding for the respective transport systems. Moreover, the genomic data was then supported with the results obtained from the API carbohydrate utilisation test. This integrated data of genomic and physiological features of the strain together provides a comprehensive outline of the mechanism of carbohydrate utilisation as depicted in Fig. 6. Our in silico analysis also revealed the presence of specialised proteolytic system, the ‘Opp proteins’, which belong to a superfamily of highly conserved ATP-binding cassette transporters that mediate the uptake of casein-derived peptides (Kunji et al. 1995; Peltoniemi et al. 2002; Vermeulen et al. 2005). This specialised machinery including the extracellular enzymes, drive the conversion of complex carbohydrates and proteins from the food to absorbable forms for the human body and thus contributing to the energy extraction in a major way.
The metabolic capacity to assimilate nitrogen and sulphur was found to be associated with the genome, along with its potential to convert nitrate to nitrite and then to reduced nitrogen such as nitrous oxide, ammonia and urea by strain 17OM39. These compounds are known to play a role in the microbiome development (Tiso and Schechter 2015). The strain 17OM39 has potential to synthesise vitamins (folate, thiamine) and essential amino acids (methionine, lysine) along with ten other amino acids. These amino acids produced serves as precursors for synthesis of short-chain fatty acids (Neis et al. 2015). Strain 17OM39 was found to be an effective reducer of cholesterol in vivo. This depletion of cholesterol in the host results in protection against cardiovascular diseases (Pereira and Gibson 2002; Ha et al. 2006).
One concern regarding the use of strains of E. faecium is that this species includes both haemolytic and non-haemolytic stains (Semedo et al. 2003b). Several are considered to have pathogenic properties. However, E. faecium is a well-known commensal and found to commonly occur in healthy individuals (Lebreton et al. 2014) Therefore, it is highly important to test for pathogenicity of E. faecium strains for safety purposes. Strain 17OM39 showed alpha haemolytic activity and was sensitive to human serum. This demonstrates the strain 17OM39 is non-pathogenic and is potentially a safe bacterium as shown by in vitro testing. Antibiotic resistance is also a major concern in strain of E. faecium (Lebreton et al. 2014) and the strain 17OM39 was sensitive to major antibiotics tested and showed only marginal sensitivity to some other antibiotic such as ciprofloxacin.
In addition, the strain 17OM39 was found to secrete an antimicrobial compound belonging to bacteriocin (NRPS group), as evident from the antimicrobial assay and genomic analysis. Moreover, the antimicrobial compound was found to be specific against L. monocytogenes. We also investigated the genome for any signatures of mobile elements, which are known to play a major role in pathogenesis (Mikalsen et al. 2015). The strain 17OM39 did not harbour any virulence genes, plasmids, pathogenic IS elements, CRISPR and Genomic Islands which are implicated to play a role in the virulence and antibiotic resistance in the bacterium (Schmidt and Hensel 2004; Gyles and Boerlin 2014; Louwen et al. 2014). Absence of genes encoding for the plasmid maintenance and transfer (oriT and tra), is an additional attribute for strain 17OM39. Based on the genome homology results, we further tried to investigate its relatedness with the marketed probiotic strain T110. It is evident from the DDH, ANI calculation and RFLP analysis (in silico analysis), that the strain 17OM39 under study is a not a clonal strain of the marketed probiotic strain 17OM39.
In summary, the integrated approach guided by genome analysis, physiological characterisation, with specific testing for probiotic potential has given crucial insights to the potential of E. faecium 17OM39 as probiotic. The strain 17OM39 fulfils all the qualities of being a probiotic and the genome insights have given more information into the molecular machinery potentially involved in the probiotic effects. Our work demonstrates the importance and usefulness of combining genome analysis with laboratory assays including in vitro testing for extensive characterisation of population-specific probiotics for the Indian population.
References
Abdhul K, Ganesh M, Shanmughapriya S, Kanagavel M, Anbarasu K, Natarajaseenivasan K (2014) Antioxidant activity of exopolysaccharide from probiotic strain Enterococcus faecium (BDU7) from Ngari. Int J Biol Macromol 70:450–454. https://doi.org/10.1016/j.ijbiomac.2014.07.026
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:1–6. https://doi.org/10.1093/nar/gkw387
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500:232–236. https://doi.org/10.1038/nature12331
Begley M, Hill C, Gahan CGM (2006) Bile salt hydrolase activity in probiotics bile salt hydrolase activity in probiotics. Appl Environ Microbiol 72:1729–1738. https://doi.org/10.1128/AEM.72.3.1729
Bhute S, Pande P, Shetty SA, Shelar R, Mane S, Kumbhare SV, Gawali A, Makhani H, Navandar M, Dhotre D, Lubree H, Agarwal D, Patil R, Ozarkar S, Ghaskadbi S, Yajnik C, Juvekar S, Makharia GK, Shouche YS (2016) Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of prevotella and megasphaera in Indian subjects. Front Microbiol 7:1–14. https://doi.org/10.3389/fmicb.2016.00660
Bikandi J, Millán RS, Rementeria A, Garaizar J (2004) In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics 20:798–799. https://doi.org/10.1093/bioinformatics/btg491
Brugiroux S, Beutler M, Pfann C, Garzetti D, Ruscheweyh HJ, Ring D, Diehl M, Herp S, Lötscher Y, Hussain S, Bunk B, Pukall R, Huson DH, Münch PC, McHardy AC, McCoy KD, Macpherson AJ, Loy A, Clavel T, Berry D, Stecher B (2016) Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol 2:16215. https://doi.org/10.1038/nmicrobiol.2016.215
Carattoli A, Zankari E, García-fernández A et al (2014) In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. 58:3895–3903. https://doi.org/10.1128/AAC.02412-14
Carrasco M, Villarreal P, Barahona S, Alcaíno J, Cifuentes V, Baeza M (2016) Screening and characterization of amylase and cellulase activities in psychrotolerant yeasts. BMC Microbiol 16:1–9. https://doi.org/10.1186/s12866-016-0640-8
Chevreux B, Wetter TSS (1999) Genome sequence assembly using trace signals and additional sequence information. Computer science and biology: proceedings of the German Conference on Bioinformatics (GCB) 99:45 – 56. Comput Sci Biol Proc Ger Conf Bioinforma 99:45–56
Chou LS, Weimer B (1999) Isolation and characterization of acid- and bile-tolerant isolates from strains of lactobacillus acidophilus. J Dairy Sci 82:23–31. https://doi.org/10.3168/jds.S0022-0302(99)75204-5
Ciszek-Lenda M, Nowak B, Śróttek M, Gamian A, Marcinkiewicz J (2011) Immunoregulatory potential of exopolysaccharide from Lactobacillus rhamnosus KL37. Effects on the production of inflammatory mediators by mouse macrophages. Int J Exp Pathol 92:382–391. https://doi.org/10.1111/j.1365-2613.2011.00788.x
Consortium THMP (2013) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. https://doi.org/10.1038/nature11234.Structure
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641. https://doi.org/10.1093/nar/27.23.4636
Evans GA (1990) Molecular cloning: a laboratory manual. Second edition. Volumes 1, 2, and 3. Current protocols in molecular biology. Volumes 1 and 2. Cell 61:17–18. https://doi.org/10.1016/0092-8674(90)90210-6
Falcony G, Joossens M, Vieira-Silva S et al (2016) Population-level analysis of gut microbiome variation. Science 352(80):560–564. https://doi.org/10.1126/science.aad3503
Forman HJ, Bernardo A, Davies KJA (2016) What is the concentration of hydrogen peroxide in blood and plasma? Arch Biochem Biophys 603:48–53. https://doi.org/10.1016/j.abb.2016.05.005
Goh YJ, Klaenhammer TR (2010) Functional roles of aggregation-promoting-like factor in stress tolerance and adherence of lactobacillus acidophilus NCFM. Appl Environ Microbiol 76:5005–5012. https://doi.org/10.1128/AEM.00030-10
Golding CG, Lamboo LL, Beniac DR, Booth TF (2016) The scanning electron microscope in microbiology and diagnosis of infectious disease. Sci Rep 6:26516. https://doi.org/10.1038/srep26516
Greene JD, Klaenhammer TR (1994) Factors involved in adherence of lactobacilli to human Caco-2 cells. Appl Environ Microbiol 60:4487–4494
Grissa I, Vergnaud G, Pourcel C (2008) CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 36:52–57. https://doi.org/10.1093/nar/gkn228
Gyles C, Boerlin P (2014) Horizontally transferred genetic elements and their role in pathogenesis of bacterial disease. Vet Pathol 51:328–340. https://doi.org/10.1177/0300985813511131
Ha C-G, Cho J-K, Lee C-H et al (2006) Cholesterol lowering effect of Lactobacillus plantarum isolated from human feces. J Microbiol Biotechnol 16:1201–1209
Halliwell B, Clement MV, Long LH (2000) Hydrogen peroxide in the human body. FEBS Lett 486:10–13. https://doi.org/10.1016/S0014-5793(00)02197-9
Hassanzadazar H, Ehsani A, Mardani K, Hesari J (2012) Investigation of antibacterial, acid and bile tolerance properties of lactobacilli isolated from Koozeh cheese. Vet Res Forum 3:181–185
Ike Y, Hashimoto H, Clewell DONB (1987) High incidence of hemolysin production by Enterococcus (streptococcus ) faecalis strains associated with human parenteral infections. High incidence of Hemolysin production by Enterococcus (streptococcus ) faecalis strains associated with human Parente. J Clin Microbiol 25:1524–1528
Jankovic I, Ventura M, Meylan V, Rouvet M, Elli M, Zink R (2003) Contribution of aggregation-promoting factor to maintenance of cell shape in Lactobacillus gasseri 4B2. J Bacteriol 185:3288–3296. https://doi.org/10.1128/JB.185.11.3288-3296.2003
Juhas M, Van Der Meer JR, Gaillard M et al (2009) Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33:376–393. https://doi.org/10.1111/j.1574-6976.2008.00136.x
Kasana RC, Salwan R, Dhar H, Dutt S, Gulati A (2008) A rapid and easy method for the detection of microbial cellulases on agar plates using Gram’s iodine. Curr Microbiol 57:503–507. https://doi.org/10.1007/s00284-008-9276-8
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J (2012) Introducing EzTaxon-e: a prokaryotic 16s rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721. https://doi.org/10.1099/ijs.0.038075-0
King LB, Swiatlo E, Swiatlo A, McDaniel LS (2009) Serum resistance and biofilm formation in clinical isolates of Acinetobacter baumannii. FEMS Immunol Med Microbiol 55:414–421. https://doi.org/10.1111/j.1574-695X.2009.00538.x
Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MWEJ, Stiekema W, Lankhorst RMK, Bron PA, Hoffer SM, Groot MNN, Kerkhoven R, de Vries M, Ursing B, de Vos WM, Siezen RJ (2003) Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci U S A 100:1990–1995. https://doi.org/10.1073/pnas.0337704100
Kunji ERS, Hagting A, De Vries CJ et al (1995) Transport of β-casein-derived peptides by the oligopeptide transport system is a crucial step in the proteolytic pathway of Lactococcus lactis. J Biol Chem 270:1569–1574
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. https://doi.org/10.1093/nar/gkm160
Lebeer S, Vanderleyden J, De Keersmaecker SCJ (2008) Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev 72:728–764. https://doi.org/10.1128/MMBR.00017-08
Lebreton F, Willems RJL, Gilmore MS (2014) Enterococcus diversity, origins in nature, and gut colonization. Enterococci from commensals to lead causes drug resist infect 1–56
Lee I, Kim YO, Park SC, Chun J (2016) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103. https://doi.org/10.1099/ijsem.0.000760
Louwen R, Staals RHJ, Endtz HP, van Baarlen P, van der Oost J (2014) The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol Mol Biol Rev 78:74–88. https://doi.org/10.1128/MMBR.00039-13
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Luo H, Zhang CT, Gao F (2014) Ori-finder 2, an integrated tool to predict replication origins in the archaeal genomes. Front Microbiol 5:1–6. https://doi.org/10.3389/fmicb.2014.00482
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39:225–229. https://doi.org/10.1093/nar/gkq1189
McInerney JO (1998) GCUA: general codon usage analysis. Bioinformatics 14:372–373. https://doi.org/10.1093/bioinformatics/14.4.372
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R (2011) AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:339–346. https://doi.org/10.1093/nar/gkr466
Mikalsen T, Pedersen T, Willems R, Coque TM, Werner G, Sadowy E, van Schaik W, Jensen LB, Sundsfjord A, Hegstad K (2015) Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics 16:282. https://doi.org/10.1186/s12864-015-1407-6
Mishra M, Nimonkar Y, Shouche YS, Kumbhare S, Vaishampayan A, Josef N, Prakash O (2015) Pantoea intestinalis sp. nov., isolated from the human gut. Int J Syst Evol Microbiol 65:3352–3358. https://doi.org/10.1099/ijsem.0.000419
Mozzi F, Gerbino E, Font De Valdez G, Torino MI (2009) Functionality of exopolysaccharides produced by lactic acid bacteria in an in vitro gastric system. J Appl Microbiol 107:56–64. https://doi.org/10.1111/j.1365-2672.2009.04182.x
Muller JA, Ross RP, Sybesma WFH, Fitzgerald GF, Stanton C (2011) Modification of the technical properties of Lactobacillus johnsonii NCC 533 by supplementing the growth medium with unsaturated fatty acids. Appl Environ Microbiol 77:6889–6898. https://doi.org/10.1128/AEM.05213-11
Neis EPJG, Dejong CHC, Rensen SS (2015) The role of microbial amino acid metabolism in host metabolism. Nutrients 7:2930–2946. https://doi.org/10.3390/nu7042930
O’Toole PW, Marchesi JR, Hill C (2017) Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol 2:17057. https://doi.org/10.1038/nmicrobiol.2017.57
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27:29–34. https://doi.org/10.1093/nar/27.1.29
Ouwehand AC, Salminen S (2003) In vitro adhesion assays for probiotics and their in vivo relevance: a review. Microb Ecol Health Dis 15:175–184. https://doi.org/10.1080/08910600310019886
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R (2014) The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res 42:206–214. https://doi.org/10.1093/nar/gkt1226
Patil VS, Salunkhe RC, Patil RH, Husseneder C, Shouche YS, Venkata Ramana V (2015) Enterobacillus tribolii gen. Nov., sp. nov., a novel member of the family Enterobacteriaceae, isolated from the gut of a red flour beetle, Tribolium castaneum. Antonie Van Leeuwenhoek 107:1207–1216. https://doi.org/10.1007/s10482-015-0412-8
Peltoniemi K, Vesanto E, Palva A (2002) Genetic characterization of an oligopeptide transport system from Lactobacillus delbrueckii subsp. bulgaricus. Arch Microbiol 177:457–467. https://doi.org/10.1007/s00203-002-0411-9
Pereira DI a, Gibson GR (2002) Cholesterol assimilation by lactic acid bacteria and Bifidobacteria isolated from the human gut. Appl Environ Microbiol 68:1–6. https://doi.org/10.1128/AEM.68.9.4689
Pieniz S, Andreazza R, Anghinoni T, Camargo F, Brandelli A (2014) Probiotic potential, antimicrobial and antioxidant activities of Enterococcus durans strain LAB18s. Food Control 37:251–256. https://doi.org/10.1016/j.foodcont.2013.09.055
Plovier H, Everard A, Druart C, Depommier C, van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, Myridakis A, Delzenne NM, Klievink J, Bhattacharjee A, van der Ark KCH, Aalvink S, Martinez LO, Dumas ME, Maiter D, Loumaye A, Hermans MP, Thissen JP, Belzer C, de Vos WM, Cani PD (2016) A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 23:107–113. https://doi.org/10.1038/nm.4236
Raghuwanshi S, Misra S, Bisen PS (2015) Indian perspective for probiotics: a review. Indian J Dairy Sci 68:3
Rastall RA, Gibson GR, Gill HS, Guarner F, Klaenhammer TR, Pot B, Reid G, Rowland IR, Sanders ME (2005) Modulation of the microbial ecology of the human colon by probiotics, prebiotics and synbiotics to enhance human health: an overview of enabling science and potential applications. FEMS Microbiol Ecol 52:145–152. https://doi.org/10.1016/j.femsec.2005.01.003
Rojo D, Celia M, Raczkowska BA et al (2016) Exploring the human microbiome from multiple perspectives: factors altering its composition and function. FEMS Microbiol Rev 63:1–26. https://doi.org/10.1093/tropej/fmw080
Saran S, Bisht MS, Singh K (2012) Comparing adhesion attributes of two isolates of Lactobacillus acidophilus for assessment of prebiotics. Int J Sci Res Publ 2:2–8
Savijoki K, Ingmer H, Varmanen P (2006) Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol 71:394–406. https://doi.org/10.1007/s00253-006-0427-1
Schmidt H, Hensel M (2004) Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev 17:14–56. https://doi.org/10.1128/CMR.17.1.14
Semedo T, Almeida Santos M, Martins P, Silva Lopes MF, Figueiredo Marques JJ, Tenreiro R, Barreto Crespo MT (2003a) Comparative study using type strains and clinical and food isolated to examine hemolytic activity and ocurrente of the cyl operon in enterococci. J Clin Microbiol 41:2569–2576. https://doi.org/10.1128/JCM.41.6.2569-2576.2003
Semedo T, Santos MA, Martins P et al (2003b) Comparative study using type strains and clinical and food isolates to examine hemolytic activity and occurrence of the cyl operon in enterococci. J Clin Microbiol 41:2569–2576. https://doi.org/10.1128/JCM.41.6.2569-2576.2003
Shen H, He X, Liu Y, Chen Y, Tang J, Guo T (2016) A complex inoculant of N2-fixing, P- and K-solubilizing bacteria from a purple soil improves the growth of kiwifruit (Actinidia chinensis) plantlets. Front Microbiol 7:1–12. https://doi.org/10.3389/fmicb.2016.00841
Shetty SA, Marathe NP, Shouche YS (2013) Opportunities and challenges for gut microbiome studies in the Indian population. Microbiome 1:24. https://doi.org/10.1186/2049-2618-1-24
Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M (2006) ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. https://doi.org/10.1093/nar/gkj014
Sridevi N, Vishwe P, Prabhune A (2009) Hypocholesteremic effect of bile salt hydrolase from Lactobacillus buchneri ATCC 4005. Food Res Int 42:516–520. https://doi.org/10.1016/j.foodres.2009.02.016
Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, Barratt MJ, VanArendonk LG, Zhang Q, Province MA, Petri Jr WA, Ahmed T, Gordon JI (2014) Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510:417–421. https://doi.org/10.1038/nature13421.Persistent
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Tatusova T, Dicuccio M, Badretdin A et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. https://doi.org/10.1093/nar/gkw569
Tiso M, Schechter AN (2015) Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS One 10:1–18. https://doi.org/10.1371/journal.pone.0119712
Tokatl M, Gülgör G, Baʇder Elmac S et al (2015) In vitro properties of potential probiotic indigenous lactic acid bacteria originating from traditional pickles. Biomed Res Int 2015:1–8. https://doi.org/10.1155/2015/315819
Vermeulen N, Pavlovic M, Matthias A et al (2005) Functional characterization of the proteolytic system of Lactobacillus sanfranciscensis DSM 20451 T during growth in sourdough functional characterization of the proteolytic system of Lactobacillus sanfranciscensis DSM 20451 T during growth in sourdough. Appl Environ Microbiol 71:6260–6266. https://doi.org/10.1128/AEM.71.10.6260
Vuye A, Pijck J (1973) Urease activity of Enterobacteriaceae: which medium to choose. Appl Microbiol 26:850–854
Whaley DN, Dowell VR, Wanderlinder LM, Lombard GL (1982) Gelatin agar medium for detecting gelatinase production by anaerobic bacteria. J Clin Microbiol 16:224–229
Whittenbury R (1964) Hydrogen peroxide formation and catalase activity in the lactic acid bacteria. J Gen Microbiol 35:13–26. https://doi.org/10.1099/00221287-35-1-13
Wu D, Sun MZ, Zhang C, Xin Y (2014) Antioxidant properties of Lactobacillus and its protecting effects to oxidative stress caco-2 cells. J Anim Plant Sci 24:1766–1771
Yusof M, Samad A, Nyonya C et al (1989) A plate assay for primary screening of lipase activity. J Microbiol Methods 9:5156–5156. https://doi.org/10.1016/0167-7012(89)90030-4
Zanotti I, Turroni F, Piemontese A, Mancabelli L, Milani C, Viappiani A, Prevedini G, Sanchez B, Margolles A, Elviri L, Franco B, van Sinderen D, Ventura M (2015) Evidence for cholesterol-lowering activity by Bifidobacterium bifidum PRL2010 through gut microbiota modulation. Appl Microbiol Biotechnol 99:6813–6829. https://doi.org/10.1007/s00253-015-6564-7
Acknowledgements
The authors are grateful to Kiran Mahale for genome analysis, and Kunal Yadav, Rakeshkumar Yadav, Aniket Saraf, and Kamlesh Jangid for critically reviewing the manuscript.
Funding
This work was supported by the Department of Biotechnology, Government of India, under Grant for National Centre for Microbial Resources (NCMR) (BT/Coord.II/01/03/2016). Additional support was provided by the Department of Biotechnology, Basaveshwar Engineering College, Bagalkot, under TEQUIP Grant to Bharati S. Meti.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure statement
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 1133 kb)
Rights and permissions
About this article

Cite this article
Ghattargi, V.C., Nimonkar, Y.S., Burse, S.A. et al. Genomic and physiological analyses of an indigenous strain, Enterococcus faecium 17OM39. Funct Integr Genomics 18, 385–399 (2018). https://doi.org/10.1007/s10142-018-0596-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-018-0596-x