
Abstract
MicroRNAs (miRNAs) are small endogenous RNAs of ~22 nucleotides that have been shown to play regulatory role by negatively affecting the expression of genes at the post-transcriptional level. Information of miRNAs on some important crops like soybean, Arabidopsis, and rice, etc. are available, but no study on heat-responsive novel miRNAs has yet been reported in wheat (Triticum aestivum L.). In the present investigation, a popular wheat cultivar HD2985 was used in small RNA library construction and Illumina HiSeq 2000 was used to perform high-throughput sequencing of the library after cluster generation; 110,896,604 and 87,743,861 reads were generated in the control (22 °C) and heat-treated (42 °C for 2 h) samples, respectively. Forty-four precursor and mature miRNAs were found in T. aestivum from miRBase v 19. The frequencies of the miRNA families varied from 2 (tae-miR1117) to 60,672 (tae-miR159b). We identify 1052 and 902 mature miRNA sequences in HD2985 control and HS-treated samples by mapping on reference draft genome of T. aestivum. Maximum identified miRNAs were located on IWGSC_CSS_3B_scaff (chromosome 3B). We could identify 53 and 46 mature miRNA in the control and HS samples and more than 516 target genes by mapping on the reference genome of Oryza sativa, Zea mays, and Sorghum bicolor. Using different pipelines and plant-specific criteria, 37 novel miRNAs were identified in the control and treated samples. Six novel miRNA were validated using qRT-PCR to be heat-responsive. A negative correlation was, however, observed between the expression of novel miRNAs and their targets. Target prediction and pathway analysis revealed their involvement in the heat stress tolerance. These novel miRNAs are new additions to miRNA database of wheat, and the regulatory network will be made use of in deciphering the mechanism of thermotolerance in wheat.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) is highly sensitive to the elevated temperatures, especially during the reproductive and grain-filling stages (Kumar et al. 2013a; Gibson and Paulsen 1999), and heat stress (HS) adversely affects the yield of the crop (Kumar et al. 2013b). It causes pollen sterility, drying of stigmatic surface, and pseudo-seed setting in wheat; shrivelled starch granules with empty pockets were observed in wheat endosperms (Barakat et al. 2007). Wide variations in the thermotolerance capacity were, however, observed among different cultivars of wheat grown under the elevated temperatures (Klevebring et al. 2009). The defense mechanism associated with tolerance against abiotic stresses has not been fully deciphered in wheat, partly because the genome of wheat is only partially sequenced. Recently, the first draft of the wheat genome has, however, been reported (Jia et al. 2013; Mayer et al. 2014). Projects are however underway to sequence the whole wheat genome under an international consortium (International Wheat Genome Sequencing Consortium; www.wheatgenome.org).
MicroRNAs (miRNAs) are small non-coding RNAs of 19 to 25 nucleotides but play important role in gene regulation in plants and animals (Voinnet 2009; Carrington and Ambros 2003); these are characterized by their precursor stem loop secondary structures and are conserved across the species (Bartel 2004; Zhai et al. 2013). Plant miRNA genes basically require protein-coding genes for their biogenesis and produce a long primary transcript (primary miRNA (pri-miRNA)) (Jia et al. 2013). Pri-miRNA is further transcribed by polymerase II and then processed by Dicer-like 1 (DCL1) into the precursor miRNA (pre-miRNA), normally of about 70–300 nucleotides (nt). The pre-miRNA is further processed into the mature miRNA: miRNA* duplex, and these processes occur in the nucleus (Wang et al. 2005; Palatnik et al. 2003; Kidner and Martienssen 2005). In the next stage, the duplex is transferred into the cytoplasm and it gets unwound (Wang et al. 2005; Palatnik et al. 2003). The miRNA is then assembled into an RNA-induced silencing complex (RISC) and guides the RISC to cleave or suppress the target messenger RNA (mRNA) (Wang et al. 2005; Palatnik et al. 2003; Khvorova et al. 2003). It also regulates the expression of gene by hybridizing to the 3′-untranslated region of mRNA or by the cleavage of mRNA. MiRNAs play major role in growth, development, and response to the environmental stresses of plants (Wang et al. 2005; Palatnik et al. 2003; Kidner and Martienssen 2005). With the identification of increasing numbers of miRNAs and their targets, our knowledge of their regulatory roles has widened over a large spectrum of plant developmental programs, including growth and developmental pattern, metabolic processes, hormone responses, stress defense, and signaling (Berezikov et al. 2006; Carthew and Sontheimer 2009). About 3171 new hairpins and 3625 novel mature products bringing the total to 21,264 have been reported in miRBase database version 19 (httpp://www.mirbase.org/cgi-bin/browse.pI); Oryza sativa, Glycine max, and Medicago sativa have the maximum identified miRNAs deposited in miRBase (Kozomara and Griffiths-Jones 2011). Next-generation high-throughput sequencing technologies open up possibilities of exploring small RNA (sRNA) populations in economically important species that lack adequate genome information such as T. aestivum. Numerous miRNAs have been identified from rice, maize, Arabidopsis, Brassica spp., pea, etc. (Rajagopalan et al. 2006; Yao et al. 2007). Ever-increasing evidence shows that the miRNA repertoire of any plant or animal species comprises of a set of conserved ancient miRNAs as well as many recently evolved species-specific miRNAs (Song et al. 2010). The availability of next-generation sequencing (NGS) technologies provide high-throughput tools to make new discoveries of additional species-specific or lowly expressed miRNAs, e.g., in Arabidopsis, rice, Poplar, Z. mays, Medicago truncatula, Lycopersicon esculentum, Gossypium hirsutum, and Taxus chinensis (Rajagopalan et al. 2006; Song et al. 2010; Meyers et al. 2008). The availability of a large number of expressed sequence tags (ESTs) from T. aestivum may also be an excellent source of experimental material for elucidation of gene expression and regulation. Although miRNA have been extensively studied in the past few years in different crops, very limited systematic study has been undertaken on the Triticum genus and especially on T. aestivum because of limited genome information (Allen et al. 2004). Sequencing of all expressed sRNAs is, however, required for complete identification of conserved and novel miRNAs in T. aestivum. Recent miRNA analysis in Arabidopsis and rice, with the deep sequencing approach discovered that the encoding loci of non-conserved miRNAs were more than that of conserved miRNAs (Carra et al. 2009); it is, therefore, necessary to extend research on the miRNAs in T. aestivum, with deep sequencing as a preferred method.
It is almost an established fact that defense mechanisms of plants against the HS depend, to a great extent, on the expression of numerous stress-associated genes (SAGs) involved in various biological pathways and miRNA act as drivers for regulating the expression of these SAGs (Fahlgren et al. 2007; Wahid et al. 2007). Identification of several miRNAs from wheat has been reported (Kurtoglu et al. 2014), but the number is very less when compared with other crops. Here, we report identification of conserved and novel heat-responsive miRNAs and their targets in wheat (T. aestivum). Validation of randomly selected miRNAs was also carried out in different tissues through quantitative real-time PCR.
Materials and methods
Plant material and stress treatment
HD2985, a popular and heat-tolerant cultivar of wheat (T. aestivum L.) was used in the present investigation and was procured from the Division of Genetics, Indian Agricultural Research Institute (IARI), New Delhi. Seeds were treated with bavistin (0.5 %) before sowing in pots. Seeds were sown in six pots (having equal quantity of perlite to FYM mixture) inside the regulated chamber (22 ± 3 °C, relative humidity of 75 %, and 8 h light with intensity of 250 μmol m−1 s−1 PAR) in the National Phytotron Facility, IARI, New Delhi. Irrigation was done at regular intervals, and plants at the pollination stage (three pots) were exposed to heat stress (HS −42 °C for 2 h), whereas other three pots served as control (22 °C). The HS was given in a sinusoidal mode using microprocessor-regulated controller with an increase of 1 °C/10 min till the temperature reaches 42 °C, and it was maintained for 2 h; further, temperature decreases to 22 °C ± 3 in the same fashion. Similarly, HD2329 (thermosusceptible) cultivar of wheat was also germinated and exposed to HS as mentioned above for the comparative analysis. Samples (root, stem, flag leaf, and pollens) were collected in triplicates from both the cultivars and freezed in liquid nitrogen for further investigation. For the validation of identified miRNAs, HD2985 and HD2329 cultivars of wheat were sown in two groups (six pots each; three for control, and three for differential HS) inside the Phytotron chamber under the regulated conditions. One group of plants (12-day-old seedlings) was exposed to HS (42 °C) for 0.5, 1, 2, 3, and 4 h for temporal expression analysis. Another group was kept at 22 °C ± 3 and was used as control. Samples were collected immediately after the treatment and stored at −80 °C for down-processing. Flowchart of the work plan has been depicted in the supplementary file (ESM_1.tif). Three biological replicates from control and HS-treated samples (HD2985) were used for the total RNA isolation and sRNA sequencing using the high-throughput platform of Illumina HiSeq 2000.
Total RNA isolation and quality analysis
One hundred milligrams of frozen tissues was ground to fine powder in liquid nitrogen using mortar and pestle, pre-chilled to −80 °C. Ground samples were transferred to RNase-free 2 mL tubes, and XcelGen kit was used for the downstream processing following the instruction of the manufacturers (Srivastava et al. 2012). The RNA was checked for quality by Bio-analyzer (Agilent, UK) and stored at −80 °C for downstream application. All the RNA samples from different tissues were mixed to form a single RNA pool. RNA pool from control and HS-treated HD2985 were used for the sRNA sequencing.
sRNA sequencing and sequence processing
Pooled RNA samples from control and HS-treated HD2985 were used for the sRNA sequencing by Xcelris India Private Limited, Ahmedabad, India, using high-throughput Illumina HiSeq (Illumina, USA). SRNAs with 16–30 nt in length were first separated from the total RNA by size fractionation with 15 % TBE urea polyacrylamide gel (TBU). The sRNA (16–30 nt in length) were excised from the gel and submerged in 600 μL of 0.3 M sodium chloride overnight at 4 °C. The gel slurry was then passed through a spin filter column (Corning, China), and RNA was precipitated by addition of 3 μL of 5 mg/mL mussel glycogen (Invitrogen, USA) and 800 μL of ethanol. The RNA pellets were then washed with 75 % ethanol and air-dried at 25 °C. The sRNA was re-suspended in 5.0 μL of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA). The isolated sRNAs were then ligated to 5′ adaptor (5′UCAGAGUUCUACAGUCCGACGAUC) using T4 RNA ligase (Promega, Madison, WI) in the presence of RNase Out (Invitrogen, UK) overnight at 4 °C according to the manufacturer’s instructions. After selecting the ligated products by size fractionation, a 3′ adaptor (5′UCGUAUGCCGUCUUCUGCUUGU) was ligated to the sRNAs following the same procedure as the ligation of the 5′ adaptor. Finally, the ligated RNAs were size fractionated on a 10 % TBE urea polyacrylamide gel, and the 70 nucleotide RNAs were excised. The 3′ adaptor ligated sRNAs were then purified from the gel and precipitated as described above followed by re-suspension in 5.0 μL DEPC-treated water (Ambion, UK). The sRNA with 5′ and 3′ adaptors were selected and reversely transcribed to complementary DNA (cDNA) with the RT primer (CAAGCAGAAGACGGCATACGA) using Superscript II reverse transcriptase (Invitrogen, UK). The cDNA was purified by 15 % TBU and dissolved in 100 μL 1 × NEB. The cDNA was quantified by Agilent-2100 and diluted to 10 nM final concentration; 18 ng cDNA was loaded into the Illumina 2000 Genome Analyzer for sequencing. The sequencing raw data of control and HS-treated samples were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the BioProject Database under the accession number PRJNA172054.
Identification and distribution pattern of conserved wheat miRNAs and their predicted targets using the reference draft genome sequence of Triticum aestivum
The raw data for HD2985 control and HS-treated samples was imported by discarding the read names and quality in CLC genomics workbench using the Illumina importer. Once the reads were imported, they were filtered for adapter sequences. While trimming the data, the minimum and maximum length of the tags was set to 15 and 55 bp. The reads having length smaller than 15 bp and greater than 55 were discarded. The next step in the analysis was to annotate the HD2985 control and HS-treated samples to identify known small RNAs. The hairpin structure of the miRNAs was predicted using the available wheat ESTs. We downloaded a total of 12,84,986 ESTs from NCBI (http://www.ncbi.nlm.nih.gov/dbEST/), and further, these ESTs were searched against Rfam database (https://www.sanger.ac.uk/resources/databases/rfam.html). First, the sequences were blasted to the Rfam database (Rfam: rRNA, tRNA, snRNA, snoRNA, and other non-coding RNAs), repeat sequences, and mRNAs. Matched sequences were discarded. The filtered reads of both the samples were mapped on the reference ESTs of wheat. The unmatched sequences were filtered. Finally, the remaining sequences were mapped to all known plant miRNA sequences to identify the conserved miRNAs in T. aestivum from the miRBase database (version 19.0, http://www.mirbase.org/ website). Closely related species (T. aestivum, Arabidopsis thaliana, S. bicolor, Z. mays, and O. sativa) were used for the annotation of miRNA using miRBase v 19. Matched sequences with no more than three mismatches were considered as candidate conserved miRNAs. At the same time, the unmatched sequences were reserved as candidate novel miRNAs. The relative abundance of miRNAs were also estimated based on the number of times each miRNA were observed to be represented in the small RNA libraries of control and HS-treated samples.
MiRNAs identified were mapped on the available reference draft genome sequence of T. aestivum (downloaded from ftp: //ftp.ensemblgenomes.org/pub/release-20/plants/fasta/triticum_aestivum/dna/. assembly version IWGSP1, Jul 2013) for distribution analysis and identification of scaffolds. The reference sequence is the collection of non-overlapping assembled sequences such that all assembled sequences are included, and each sequence region is included in the largest possible assembly. It comprises of 709,031 scaffolds with 4,419,109,344 bp. The 91,266,903 and 21,175,164 trimmed reads were obtained from a total of 110,896,604 and 21,284,027 for HD2985 control and HS-treated samples with an average read length from 22 to 26 bp, respectively. MiRBase v 19 and known non-coding RNAs (Rfam database) were used for the annotation. Identified scaffolds showing the alignment with candidate microRNAs were considered for the target prediction using the web-based tool psRNA Target program (http://plantgrn.noble.org/psRNATarget/?function=3) with default parameters. The program uses a scale of 0–5 to indicate the stringency of miRNA-target pairing; the smaller numbers represent higher stringency. Score of 3 was used in this analysis. Parameters used for psRNA Target program was (a) maximum expectation—3.0, (b) length for complementarily scoring (hspsize)—20, (c) target accessibility—allowed maximum energy to unpair the target site (UPE)—≤25, and (d) range of central mismatch leading to translational inhibition—9–11 nt. Novel and conserved candidate miRNA sequences were blasted against drafted T. aestivum genome sequences, and their flanking sequences in the genome were used to predict their secondary structures by using the mfold web server (http://mfold.rna.albany.edu/?q=mfold/download-mfold website).
Differential expression analysis of identified mature miRNAs
The fragments per kilobase of exon per million fragments mapped (FPKM) values for each mature miRNA identified from control and HS-treated samples were calculated and were used to assess the log fold change as Log2 (FPKM_HD2985_treated/FPKM_HD2985_control). Then, p value for the chi-square (χ 2) test statistic for each mature miRNA was calculated with the following formula:
where x = reads mapped on the control sample, n = total reads mapped on miRBase in the control sample, y = reads mapped on treated sample, m = total reads mapped on miRBase in treated sample. Based on the p values, statistically significant (p value less than or equal to 0.05) miRNAs were identified and were categorized as upregulated and downregulated based on log fold change.
Novel miRNA identification
In order to streamline our study based on homology, O. sativa, Z. mays, and S. bicolor were selected as closest homologs based on the taxonomic information to identify conserved and novel miRNAs in the control and HS-treated samples of wheat (HD2985). The reference genomes of O. sativa build MSU6.1 and Z. mays build B73_RefGen_v2 were downloaded from plantGDB database and that of S. bicolor build JGI_sbi1 from JGI. There were 581, 172, and 171 known precursor miRNAs and 661, 321, and 172 known mature miRNA sequences in hairpin.fa and mature.fa respectively for O. sativa, Z. mays, and S. bicolor. The filtered reads from the control and HS-treated samples were mapped on to the reference genome and miRBase precursor and mature sequences of O. sativa, Z. mays, and S. bicolor. MiRanalyzer pipeline was used to predict the potential T. aestivum miRNA precursor molecules based on homology. The work-flow chart has been depicted in the supplementary file (ESM_2.tif). The precursor molecules were extracted from the genome, and secondary structure prediction was carried out using the mfold web server (http://mfold.rna.albany.edu/?q=mfold). Further, selection of novel precursor from the MiRanalyzer (http://bioinfo5.ugr.es/miRanalyzer/miRanalyzer.php) predicted candidate precursor molecules was done using Xcelris proprietary miRNA pipeline (Xcelris Labs, India). After applying the above criteria, the predicted candidate miRNA precursor molecules were filtered using miRCheck pipeline (Bartel Lab, USA) based on following plant-specific criteria:
-
All predicted base pairs must be in the same direction (i.e., all nucleotides pairing to the mature miRNA must either all be 5′ of the mature miRNA or all be 3′ of the mature miRNA).
-
No more than four nucleotides may be unpaired, with no more than two adjacent unpaired nucleotides.
-
The length of the hairpin, measured as the number of nucleotides containing the mature miRNA, the loop, and the nucleotides predicted to pair to the mature miRNA (including an equal number of terminal unpaired nucleotide, if any) must be at least 60.
-
No more than one nucleotide in mature miRNA may be asymmetrically unpaired.
-
The pairing must extend three nucleotides beyond the mature miRNA. In addition, the extended mature miRNA* (here defined as the sequence base pairing to the extended mature miRNA, including an equal number of terminal unpaired nucleotide, if any), must not be longer than 27 nucleotides, with no more than 5 nucleotides unpaired in total and no more than 3 adjacent unpaired nucleotides.
-
Finally, at least one nucleotide in either the extended mature miRNA or extended mature miRNA* must be unpaired.
The structure of novel miRNA was also analyzed for minimum folding free energy index (MFEI), and structures having value greater than or equal to 0.85 were selected. The following equations were used to determine MFEI:
Prediction of potential target mRNA for identified miRNAs
The predicted target sites of miRNA candidates were identified by aligning the miRNA sequences with the assembled ESTs of T. aestivum using the psRNA Target program with default parameters (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao 2011). All predicted target genes were evaluated by scoring, and the criteria used were the following: each G:U wobble pairing was assigned 0.5 point; each indel was assigned 2.0 points; and all other non-canonical Watson-Crick pairings were each assigned 1.0 point. The total score for an alignment was calculated on the basis of 20 nt; when the query was longer than 20 nt, scores for all possible consecutive 20 nt subsequence were computed, and the minimum score was considered to be the total score for the query-subject alignment. Because targets complementary to the miRNA 5′ end appear to be critical, mismatches other than G:U18 wobbles at positions 2–7 at the 5′ end were further penalized by 0.5 point in the final score. Sequences were considered to be miRNA targets if the total score was less than 3.0 points (Zhao et al. 2010). For the identification of miRNA targets involved in thermotolerance pathways, we retrieved the mRNA transcript sequences from the public biological databases based on the literature search. We downloaded 465 thermotolerant plant mRNA transcript sequences in FASTA format and submitted in the KEGG Automatic Annotation Server (KAAS) for the identification of pathways (http://www.genome.jp/kegg/pathway.html).
Validation of miRNAs and their targets using quantitative real-time PCR
Designing of primers
A mature miRNA-specific forward primer was designed using Genefisher2 primer designing software (http://bibiserv.techfak.uni-bielefeld.de/genefisher2/), and quality was checked using Oligo Analyzer (Integrated DNA Technologies, USA; Table 1). Seven forward primers were designed based on the mature miRNA sequences. In case Tm of a mature miRNA was <60 °C, it was adjusted by adding Gs or Cs to the 5′ end and/or As to the 3′ end of the miRNA sequence. U6 snRNA (Clontech, USA) was used as endogenous control to normalize the data.
For comparative expression analysis of miRNAs and their target genes, eight conserved mature miRNAs and their respective target genes were randomly selected. Primers for miRNAs were designed as explained in the above section, whereas target gene-specific forward and reverse primers were designed using Genefisher2 primer designing software (http://bibiserv.techfak.uni-bielefeld.de/genefisher2/). The quality of the primers was checked using Oligo Analyzer. The list of primers used for the comparative expression analysis has been presented in Online Resource 3 (ESM_3.xls).
Quantitative real-time PCR
The validation of identified miRNAs and their targets in the present investigation was carried out using the quantitative real-time PCR expression analysis. The validation was carried out in two contrasting wheat cultivars—HD2985 (thermotolerant) and HD2329 (thermosusceptible); both are popular cultivars in southeast Asia. A new approach was followed for the expression profiling of miRNAs. MiRNAs were isolated from the control and HS-treated samples of both the cultivars using mirVana™ miRNA Isolation Kit (Ambion, USA). The quality of isolated miRNA was checked on Bio-analyzer (Agilent), and we observed OD 260/280 ratio of >2.0. The isolated miRNAs were also analyzed on the 15 % TBE acrylamide gels, and prominent RNA bands were observed with ethidium bromide staining. The first-strand cDNA synthesis was carried out using the Mir-X™ miRNA First-Strand Synthesis kit (Clontech, USA) following the manufacturer’s instruction. In brief, 2.5 μg of total RNA was polyadenylated with ATP by poly (A) polymerase. The poly (A)-tailed total RNA was reverse-transcribed by PrimeScript® RTase by using a universal adapter primer (containing oligo-dT). Quantitative RT-PCR analysis was carried out by using SYBR® Premix Ex TaqTM II (Perfect Real Time) (TaKaRa, Japan) on a Bio-Rad CFX96 machine. All reactions were performed in triplicate for each sample, and U6 SnRNA (Clontech, USA) was used as the internal control gene. Relative expression levels of miRNAs were quantified by using the 2−ΔΔCt method (pfaffl 2001). For target genes, first-strand cDNA synthesis was carried out using RevertAidTM H minus First-Strand cDNA synthesis kit (Thermo Scientific, USA), and quantitative real-time PCR was performed on CFX96 platform (Bio Rad, UK) using the SYBR Advantage® qPCR Premix (Clontech, USA).
Results
Identification and distribution pattern of conserved mature miRNAs and their predicted targets using the reference draft genome sequence of Triticum aestivum
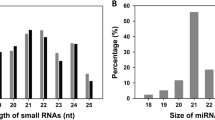
In recent years, numerous diverse roles of miRNAs have been unraveled for growth, development, and defense, and the evolutionary conservation of miRNA families has provided simple and efficient approach to identify conserved miRNAs in different plant species. Here, we relied on wheat EST sequences to predict hairpin structures on the basis of miRNA surrounding sequences. Wheat ESTs were used as a reference to map filtered reads resulting in 20,058,139 and 11,752,696 reads for the control and the HS-treated samples. The reads were aligned individually against precursor and mature miRNA sequences from all the plants present in miRBase v 19 resulting into alignment of 80,956 and 1,175,285 reads against mature miRNAs and 14,757 and 33,421 reads on precursors for the control and HS-treated samples, respectively. The reads mapped on mature miRNAs were used for the expression profiling. A total of 44 known precursor miRNAs and 44 mature miRNA sequences in hairpin.fa and mature.fa respectively for T. aestivum are present in miRBase v 19. The expression of tae-miR5084 was observed exclusively in the control, whereas expression of tae-miR1130, tae-miR1136, tae-miR395a, and tae-miR408 was observed only in the HS-treated sample; expression of 14 miRNAs was observed in both the control as well as HS-treated samples, whereas the expression of 25 conserved miRNAs was neither detected in the control nor in the HS-treated samples (Table 2). Here, we observed variations in the expression of 19 miRNAs in control and HS-treated samples out of 44 identified miRNAs.
The number of times each miRNA is represented in the small RNA library could serve as an index for the estimation of the relative abundance of miRNA. The large number of miRNA sequences generated in this study was used to determine their relative abundance in wheat (T. aestivum). The frequencies of the miRNA families varied from 2 (tae-miR1117, tae-miR171b, tae-miR395a) to 60,672 (tae-miR159b), indicating that expression greatly varies among the different miRNA families in wheat (Fig. 1); 422 and 357 unique miRNAs were identified in filtered data (control and HS-treated samples) mapped against all plants in miRBase v 19. List of miRNAs identified in control and HS-treated samples of HD2985 cultivar using the reference genome of T. aestivum has been attached as Online Resource 4 (ESM_4.xls).

The frequency of conserved mature miRNAs present in the sequenced small RNA library of control and heat shock-treated samples of HD2985 cultivar of wheat (Triticum aestivum L.); next-generation sequencing was performed using Illumina HiSeq 2000 platform
Homology-based miRNA identification using the reference sequence of T. aestivum (comprises of 709,031 scaffolds with 4,419,109,344 bp) and annotation using miRBase v 19 and known non-coding RNAs (Rfam database) resulted in identification of 1052 and 902 mature miRNA sequences for HD2985 control and HS-treated samples. Species wise distribution analysis of identified mature miRNAs in control sample of HD2985 showed 58, 25, 14, 2, and 1 % of the miRNA from T. aestivum, Z. mays, O. sativa, A. thalianaz, and Vitis vinifera. Similarly, in the case of HS-treated sample, we observed 42, 42, 11, 2, 2, and 1 % of the miRNA from T. aestivum, Z. mays, O. sativa, A. thaliana, V. vinifera, and S. bicolor. Maximum miRNAs were identified from T. aestivum followed by Z. mays and O. sativa in both the samples. MiR1136 identified from T. aestivum showed maximum expression level (absolute count) in both the control and HS-treated samples (Table 3). Similarly, tae-miR117 showed high expression (219 reads) in control compared to HS-treated sample (88 reads). MiR166k, miR166j, and miR166n identified from Z. mays showed very high expression in HS-treated sample (117 reads) followed by miR159a, miR159b, miR159f, miR159t, and miR159k (102 reads). The absolute count (expression level) was observed maximum in control and HS-treated samples of T. aestivum followed by Z. mays.
In the present investigation, we identified 169 miRNAs (132 miRNA by homology-based approach and 37 novel miRNAs using different analyzers and plant-specific parameters). Out of 169 miRNA, 168 got mapped on the reference draft genome sequence of wheat (Mayer et al. 2014). The distribution of miRNAs plotted on different chromosomes is shown in Fig. 2. It is evident from the figure that maximum number of miRNAs (22 miRNAs) is mapped on IWGSC_CSS_3B_scaff followed by IWGSC_CSS_5BL_scaff (14 miRNAs) and IWGSC_CSS_2BL_scaff (10 miRNAs). Five miRNAs each were mapped on IWGSC_CSS_2DL_scaff, IWGSC_CSS_4DS_scaff, IWGSC_CSS_5AS_scaff, IWGSC_CSS_6AL_scaff, IWGSC_CSS_7BL_scaff, and IWGSC_CSS_7DL_scaff. Similarly, one miRNA each was mapped on six scaffolds (IWGSC_CSS_2DS_scaff, IWGSC_CSS_3DL_scaff, IWGSC_CSS_4BS_scaff, IWGSC_CSS_4DL_scaff, IWGSC_CSS_6BS_scaff, and IWGSC_CSS_7AS_scaff). Other scaffolds identified showed the alignment with two, three, or four miRNAs each as depicted in the Fig. 2. It is well established that one chromosome may be represented by many scaffolds or single scaffold depending on how well the genome has been reconstructed or assembled from the available reads. In case of T. aestivum, recently, only the first draft of the survey sequence has been submitted on public domain by the International Wheat Genome Sequencing Consortium (IWGSC) (Mayer et al. 2014). Out of 168 identified miRNAs, 22 miRNAs were observed to be localized on the chromosome 3B, 5 miRNA on chr 5B, and 10 miRNA on chr 2B (Fig. 3). Similarly, nine and eight miRNAs were mapped on chr 6D and chr 7A. Six miRNAs were observed on each of the chr 5A, chr 6A, chr 6B, and chr 6D and 5 miRNAs on chr 2D, chr 4D, chr 5A, chr 6A, chr 7B, and chr 7D. Maximum number of miRNAs were mapped on chr 3B and minimum on chr 3D (Fig. 3). The 37 candidate microRNAs from HD2985 control and HS treated were mapped on the reference draft genome sequence of T. aestivum and showed alignment with 27 scaffolds which was further used for the target prediction. For example candidate_4832 was mapped on IWGSC_CSS_2AL_scaff, IWGSC_CSS_2BL_scaff, IWGSC_CSS_2BS_scaff, IWGSC_CSS_2DL_scaff, IWGSC_CSS_3B_scaff, and IWGSC_CSS_6DS_scaff (Table 4). Total of 41 unique targets were predicted from 7 candidate miRNAs out of 37. The maximum targets (10) were identified for candidate_4832 and candidate_6660 (Table 4). Some of the targets identified are peptide transporter PTR2 (Aegilops tauschii), wall-associated receptor kinase 4 (Triticum urartu), putative LRR receptor-like serine/threonine-protein kinase (A. tauschii), serine carboxypeptidase-like 18 (Triticum urartu), luminal-binding protein 2 (A. tauschii), etc. A list of candidate miRNAs, identified targets and their functional annotation based on the reference draft genome of T. aestivum (retrieved from ftp://ftp.ensemblgenomes.org), has been presented as Online Resource 5 (ESM_5.xls).

Distribution of miRNAs identified based on the reference drafted genome sequence of Triticum aestivum downloaded from ftp://ftp.ensemblgenomes.org/pub/release-20/plants/fasta/triticum_aestivum/dna/. assembly version IWGSP1, Jul 2013; 169 miRNAs identified in the present investigation (132 miRNA were identified by homology-based approach and 37 novel miRNAs based on different pipeline analyzers) were mapped on the drafted genome sequence of wheat

Chromosomal localization of identified miRNAs based on the survey sequence of Triticum aestivum downloaded from ftp://ftp.ensemblgenomes.org/pub/release-20/plants/fasta/triticum_aestivum/dna/. assembly version IWGSP1, Jul 2013; 168 miRNAs identified in the present investigation were mapped on the drafted genome sequence of wheat
Differential expression of mature miRNA
The FPKM value for each mature miRNA was calculated, and based on the p values, 10 statistically significant (p value less than or equal to 0.05) miRNAs were identified and were categorized as upregulated and downregulated based on log fold change. Tae-miR156, tae-miR167, tae-miR395b, and tae-miR398 were observed to be upregulated in both the control as well as HS-treated samples. The maximum log fold change in the expression was observed in tae-miR156 (3.99-fold) followed by tae-miR395b (2.4-fold) under the HS treatment. Downregulation of tae-miR1117, tae-miR159a, tae-miR159b, tae-miR160, tae-miR171a, and tae-miR319 were observed under HS in HD2985 cultivar of wheat (Table 5). Maximum downregulation was observed in the case of tae-miR319 (−4.9-fold).
Homology-based identification and distribution of wheat miRNA using the reference genome of closely related plant species
Based on the taxonomic information, we selected O. sativa, Z. mays, and S. bicolor as closest homologs for the identification of conserved and novel miRNAs in the control and HS-treated samples of wheat. We observed 30, 14, and 9 mature miRNAs in the control and 25, 10, and 11 mature miRNAs in the HS-treated samples based on the reference genome of O. sativa (build MSU6.1), Z. mays (build B73_RefGen_v2), and S. bicolor (build JGI_sbi1), respectively. The distribution of identified miRNA expression between the control and HS-treated samples were also estimated. We observed 12, 5, and 5 mature miRNAs to be exclusively expressed in the HS treated, compared to 17, 9, and 3 mature miRNAs in the control based on the reference genome of O. sativa, Z. mays, and S. bicolor (Fig. 4). Similarly, 13 (O. sativa), 5 (Z. mays), and 6 (S. bicolor) miRNAs were observed to be expressed in both the control and HS-treated samples of HD2985 cultivar of wheat. Expression of 619 (O. sativa), 302 (Z. mays), and 158 (S. bicolor) miRNAs were not detected in control and HS-treated samples. List of miRNAs identified in control and HS-treated samples of HD2985 (thermotolerant) cultivar of wheat based on homology search using the reference genome of rice, maize, and sorghum has been presented as Online Resource 6, 7, and 8 (ESM_6.xls, ESM_7.xls, ESM_8.xls).

Distribution analysis of mature miRNAs identified based on homology search using the reference genome of Oryza sativa, Zea mays, and Sorghum bicolor in control and heat stress-treated samples of HD2985 cultivar of wheat (Triticum aestivum); reference genomes of O. sativa build MSU6.1 and Z. mays build B73_RefGen_v2 were downloaded from plantGDB database and S. bicolor build JGI_sbi1 from JGI
Differential expression of mature miRNAs identified based on reference genome search
We used the FPKM value of 19 (T. aestivum), 19 (O. sativa), 19 (Z. mays), and 14 (S. bicolor) miRNAs to calculate the log fold change as Log2 (FPKM_HD2985_treated/FPKM_HD2985_control). Based on the p value, 10, 7, 1, and 2 statistically significant (p value less than or equal to 0.05) miRNAs were identified (using the reference genome of T. aestivum, O. sativa, Z. mays, and S. bicolor) and were categorized as upregulated or downregulated based on the log fold change. We observed 4, 6 (T. aestivum), 4, 3 (O. sativa), 1 (Z. mays), and 1, 1 (S. bicolor) significantly upregulated and downregulated mature miRNAs based on the reference genome search. Significant upregulation were observed in osa-miR2925 (4.04-fold), zma-miR166a (0.05-fold), sbi-miR398 (3.29-fold) and downregulation in case of osa-miR396c (−2.14-fold), sbi-miR159 (−0.039-fold), etc.
Target gene prediction
Since plant miRNA have near perfect complementarity to their targets, computational prediction has proved to be effective to identify the targets. In plants, the miRNA target sites are predominantly in the coding region. Interestingly, 187 genes were targeted by 19 miRNAs in wheat which are perfectly complementary to each other (Fig. 5). The tae-miR156 is involved in regulating squamosal promoter binding protein gene (SBP) in different crops. Similarly, tae-miR159a regulates the expression of MYB3 (transcriptional activator), alkaline phosphatase family protein (ALPL), cytochrome P450 (CYP450), cobalamine adenosyl transferase (cobO), Mob1-like protein and TLD family protein, etc. Tae-miR159b is involved in regulating target genes like MYB3 transcription factor, ubiquitin carboxyl terminal hydrolase (UCHL1; NB-ARC domain containing protein), etc. Tae-miR160 plays a very important role in plant under the abiotic stresses, by regulating the expression of target genes like auxin response factor (ARF), tetratricopeptide repeat (TPR), etc. The identified miRNAs from the constructed libraries of the control and HS-treated HD2985 cultivar of wheat were also observed to regulate the signaling pathways involved in modulating the defense mechanism under the HS. Tae-miR164 and sbi-miR164c were observed to regulate the expression of target genes involved in mitogen-activated protein kinase (MAPK) signaling pathways; it also regulate the transcript of NAC transcription factor. Tae-miR319 and sbi-miR1435b were observed to regulate MYB3 and MYB27 gene families as well as histone protein-associated genes. Superoxide dismutase (SOD) gene family was observed to be regulated by tae-miR398 and sbi-miR38. Similarly, osa-miR3979–5 was observed to have unique stress protein/transcription factor gene family as their specific targets. Osa-miR529-b and sbi-miR396c were observed to regulate the expression of ethylene responsive factor (ERF) and HSP81-1 genes. List of miRNAs (identified using the reference genome of closely related species) and their specific targets have been presented in Online Resource 9 (ESM_9.xls).

Distribution of gene targets of identified conserved miRNAs from Triticum aestivum; 187 genes were targeted by 19 miRNAs
Our investigation in other plant species showed 308, 138, and 70 target genes in O. sativa (osa), Z. mays (zma), and S. bicolor (sbi) regulated by 37, 8, and 13 identified miRNAs, respectively (Fig. 6). In case of O. sativa, we observed maximum targets associated with osa-miR2122, osa-miR1439 (25 targets), and osa-miR1436 (24 targets). Osa-miR2122 has been predicted to regulate putative transposon protein, whereas osa-miR1439, zma-miR166a, and zma-miR827 have maximum targets (30 and 29) compared to other identified miRNAs expressed in Z. mays. In S. bicolor, the maximum gene targets were observed to be associated with sbi-miR296 and sbi-miR328, each having nine targets.

Distribution of identified miRNAs based on homology search using the reference genome of O. sativa, Z. mays, and S. bicolor and their targets; Triticum aestivum—tae, Oryza sativa—osa, Zea mays—zma, Sorghum bicolor—sbi
Novel miRNA discovery using the reference genome of related plant species
The number of miRNAs identified from T. aestivum is very less compared with other closely related plant species as very few miRNAs have been identified from T. aestivum and submitted in the repository till date. Now, onus is on the discovery of novel heat-responsive miRNAs from wheat and to use it for deciphering the mechanisms associated with abiotic/biotic stress tolerance.
Novel miRNA identified based on the reference genome of Oryza sativa
Novel miRNA identification was carried out in the control and HS-treated HD2985 cultivar of wheat using an input reads of 87,160,626 and 71,104,328. The reference genome of O. sativa build MSU6.1 was downloaded from plantGDB database. Based on MiRanalyzer analysis, we observed 7889 (control) and 6848 (HS treated) precursor miRNAs in O. sativa. Using the Xcelris proprietary miRNA pipeline and miRCheck, the number of predicted precursor miRNAs was reduced to 16 and 2 in the control and HS-treated samples, respectively (Table 6). Finally, based on MFEI calculation, the novel miRNAs predicted was 16 (control) and 2 (HS treated) using the reference genome of O. sativa (Fig. 7).

Histogram showing the number of novel miRNAs predicted in control and heat shock-treated samples of HD2985 cultivar based on homology search using the reference genome of Oryza sativa, Zea mays, and Sorghum bicolor
Novel miRNA identified based on the reference genome of Zea mays
The reference genome of Z. mays build B73_RefGen_v2 was downloaded from plantGDB database. We identify 9722 (control) and 8711 (HS treated) potential candidate novel miRNAs based on MiRanalyzer predicted precursor. Further, we were able to identify 48 and 39 miRNAs in the control and HS-treated samples using Xcelris proprietary miRNA pipeline. The predicted candidate miRNA precursor molecules were further subjected to MiRCheck pipeline (using plant-specific criteria as mentioned in the earlier section) for the prediction of novel miRNA. We observed eight and nine novel heat-responsive miRNAs in the control and HS-treated samples based on miRCheck pipeline and MFEI calculation (Table 6; Fig. 7).
Novel miRNA identified based on the reference genome of Sorghum bicolor
The reference genome of S. bicolor build JGI_sbi1 was downloaded from JGI Genome portal. Based on the mapping of input reads on the reference genome of S. bicolor and processing of data using MiRanalyzer pipeline, we observed 1213 and 1157 miRNAs in the control and HS-treated samples, respectively. Further, the information was down-processed using Xcelris Proprietary Script-based pipeline, and we observed five miRNAs in both control as well as HS-treated samples. The candidate precursor miRNAs were processed using MiRCheck pipeline, and one miRNA precursor was observed in both control as well as HS-treated samples (Table 6). The predicted precursor miRNAs were further evaluated using MFEI calculation, and finally, one novel heat-responsive miRNA was identified in both the control as well as HS-treated samples of HD2985 cultivar of wheat (Fig. 7).
Based on our NGS input sequences from the control and HS-treated samples and their mapping on reference genomes of related plant species like O. sativa, Z. mays, and S. bicolor, we have identified 18, 17, and 2 novel miRNAs using different pipeline analyzers and MFEI values.
Validation of novel miRNAs for heat-responsive nature by quantitative real-time PCR
The expression pattern of novel miRNAs was studied in wheat exposed to HS in order to validate as well as predict their functions. To get an idea about the possible stage- or tissue/organ-dependent roles of miRNAs, we examined the expression patterns of miRNAs in different tissues like roots, stem, and leaves of HD2985 (thermotolerant) and HD2329 (thermosusceptible) cultivars of wheat at the seedling stage (Fig. 8). Seven novel miRNAs (candidate_430, candidate_3466, candidate_5064, candidate_5652, candidate_5993, candidate_3182, and candidate_6941) were randomly selected for the validation by quantitative real-time PCR (qRT-PCR). The list of miRNA-specific forward primers used for the qRT-PCR has been depicted in Table 7. The expression of miRNA was analyzed using the poly (T) adaptor RT-PCR method.

Validation of identified novel miRNAs (candidate_6941, candidate_3182, candidate_5652, candidate_3466, candidate_430, and candidate_5064) in root, stem, and leaves of thermotolerant (HD2985) and thermosusceptible (HD2329) wheat cultivars by quantitative real-time PCR (qRT-PCR); expression was not observed in case of candidate_5993; C—22 °C, T—42 °C for 2 h. Variation in the expression under heat stress was observed in six miRNAs out of seven miRNAs randomly selected. The expression of U6 SnRNA was used as endogenous control for normalizing the data. Relative expression was calculated by Pfaffl method
Candidate_6941 showed significant downregulation in response to HS in root, stem, and leaves of HD2985 cultivar of wheat (Fig. 8). The expression was very low in leaf compared to stem and root under HS. Similarly, candidate_3182, candidate_5652, and candidate_430 showed significant downregulation in root, stem, and leaf under the HS. The expression of candidate_430 and candidate_5652 was highly downregulated in leaves compared to other tissues in response to HS. Very low transcripts of candidate_430 were observed in root, stem, and leaves under HS. Candidate_3466 and candidate_5064 showed upregulation in response to HS in root, stem, and leaves of HD2985 cultivar of wheat. The transcripts of both the candidate miRNAs were observed abundant in root followed by stem, and minimum was observed in leaves under HS.
Validation of identified candidate miRNAs was also carried out in contrasting wheat cultivar (HD2329; thermosusceptible) in order to know the variations in the pattern of expression of miRNAs under HS in thermotolerant and thermosusceptible cultivars (Fig. 8). Candidate_6941, candidate_3182, candidate_430, and candidate_5064 showed downregulation under HS in root, stem, and leaves of HD2329 cultivar of wheat. The variations among the tissues in response to HS were non-significant in thermosusceptible cultivar. Similarly, candidate_5652 and candidate_3466 showed upregulation in response to HS in root, stem, and leaves. Relative expression of candidate_3466 was observed maximum in leaves, and candidate_5652 showed maximum expression in stem. We observed downregulation of candidate_5652 in leaves of HD2329 and upregulation in leaves of HD2985 under HS (Fig. 8).
The expression of candidate_5993 was not observed in any of the tissues of HD2985 and HD2329 under HS. Out of seven selected miRNAs, we observed significant variations in the expression of six miRNAs (candidate_430, candidate_3466, candidate_5064, candidate_5652, candidate_3182, and candidate_6941). It is the variation in the expression under HS which validates the heat-responsive nature of these identified candidate miRNAs. These heat-responsive miRNAs are predicted to regulate the expression of SAGs (their targets) which, in turn, modulate the defense mechanism of wheat under the HS.
Chromosomal localization and structural analysis of identified novel miRNAs
The identified novel miRNAs were also characterized for their chromosomal localization using the miRNA_clusters_by Chr.pl written in Pearl Script language (Table 6). The program was used to determine miRNA clusters on each chromosome given a maximum inter-miRNA distance and an input file of miRNA locations. Most of the miRNAs identified based on the reference genome of O. sativa were observed to be localized on chromosome 2 (four miRNAs) followed by chr. 7 and chr. 8 (three miRNAs each). The structural analysis of identified heat-responsive miRNAs was also carried out using Flicker v3.0 (Illumina, USA). Mapping to mature and precursor miRNA showed significant variations in the internal structure of identified novel miRNAs. The structural analysis were performed using a modified Zuker’s algorithm and were then compared to the structures derived using mfold (Zuker 1989). Variations were observed in the secondary structure of the identified heat-responsive miRNAs from wheat. The secondary structure of identified novel miRNAs has been presented in the supplementary file (ESM_10.tif).
Temporal expression analysis of novel heat-responsive miRNAs in wheat under the heat stress
Three randomly selected novel heat-responsive miRNAs (candidate_3182, candidate_3466, and candidate_430) were used for the temporal expression analysis in root, stem, and leaves of HD2985 and HD2329 (contrasting wheat cultivars) after exposing the plants to HS of 42 °C for 0.5, 1, 2, 3, and 4 h. Relative expression of candidate_3182 in roots of HD2985 showed significant upregulation in response to HS for 0.5 h, whereas further downregulation was observed under HS for 1, 2, 3, and 4 h (Fig. 9). Maximum downregulation of candidate_3182 was observed under HS for 2 h. Similarly, relative expression of candidate_3182 in roots of HD2329 showed significant downregulation in response to HS for different durations. Maximum downregulation was observed in response to HS for 3 h. Non-significant increase in the expression of candidate_3182 was observed in response to HS duration beyond 2 h (HD2985) and 3 h (HD2329), respectively.

Temporal expression analysis of identified heat-responsive miRNA (candidate_3182) in root, stem, and leaves of HD2985 (thermotolerant) and HD2329 (thermosusceptible) cultivars of wheat (Triticum aestivum L.), Plants were exposed to heat stress of 42 °C for 0.5, 1, 2, 3, and 4 h, The expression of U6 SnRNA was used for normalizing the data, Relative expression was calculated by Pfaffl method
Relative expression of candidate_3182 in stem of HD2985 showed similar pattern of downregulation as observed in root; non-significant differences were observed in response to HS for 2, 3, and 4 h (Fig. 9). In HD2329, the maximum downregulation was observed in response to HS for 1 h; variations between the treatments were non-significant. Expression of candidate_3182 in leaves of HD2985 showed significant variations among treatments. We observed maximum downregulation in response to HS for 1 h, and further increase in the expression was observed with increase in the duration of HS (Fig. 9). A non-significant change in the expression was observed in the leaves of HD2329 in response to different treatments.
Relative expression of candidate_3466 in roots of HD2985 showed upregulation under HS for 0.5 to 2 h, and further downregulation was observed with increase in the duration of HS (Fig. 10). In case of roots of HD2329, the relative expression of candidate_3466 showed significant upregulation in response to the treatments. Transcript profiling in stem and leaves showed upregulation of candidate_3466 in both the cultivars; the expression was observed more in leaves compared to stem. Similarly, we observed decrease in the relative expression of candidate_430 in roots of both the cultivars with increase in the duration of HS; non-significant variations were observed in response to treatments (Fig. 11). Maximum downregulation was observed in response to HS for 4 h in both the cultivars. The relative expression of candidate_430 in stem of HD2985 showed significant downregulation in response to the treatments. Similarly, we observed significant downregulation of candidate_430 in stems of HD2329 with increase in the duration of HS; expression was highly downregulated under HS for 4 h. The relative expression of candidate_430 was observed more in leaves of HD2329 compared to HD2985 under HS; decrease in the transcripts were observed with increase in the duration of HS in both the cultivars (Fig. 11).

Temporal expression analysis of validated heat-responsive miRNA (candidate_3466) in root, stem, and leaves of HD2985 (thermotolerant) and HD2329 (thermosusceptible) cultivars of wheat (Triticum aestivum L.). Plants were exposed to heat stress of 42 °C for 0.5, 1, 2, 3, and 4 h. The expression of U6 SnRNA was used for normalizing the data. Relative expression was calculated by Pfaffl method

Temporal expression analysis of candidate_430 in root, stem, and leaves of HD2985 (thermotolerant) and HD2329 (thermosusceptible) cultivars of wheat (Triticum aestivum) in response to heat stress of 42 °C for 0.5, 1, 2, 3, and 4 h. The expression of U6 SnRNA was used as endogenous control for normalizing the data. Relative expression was calculated by Pfaffl method
Prediction of gene targets of novel heat-responsive miRNAs
For greater understanding of the functions of newly identified heat-responsive novel miRNAs (25 and 12 for control and HS-treated samples), targets of these miRNAs were predicted in T. aestivum, O. sativa, S. bicolor, and Z. mays. The number of target genes identified for the novel miRNAs in control and HS-treated samples were 83, 26 (T. aestivum), 109, 23 (O. sativa), 82, 153 (Z. mays), and 50, 12 (S. bicolor) (Table 8). Based on the reference draft genome sequence of T. aestivum, we were able to identify target genes like peroxidase, receptor protein kinase, NADPH-cytochrome P450 reductase, Rubisco, etc. for miRNAs identified in the control and HS-treated samples. Similarly, we observed CDPK, bZIP, MYB, etc. as targets for the miRNAs identified using O. sativa as reference genome. The predicted novel miRNA targets based on the homology search using the reference genome of O. sativa, Z. mays, and S. bicolor has been attached as Online Resource 11 and 12 (ESM_11.xls, ESM_12.xls).
Identification of miRNA targets involved in thermotolerance pathways
We downloaded 465 plant mRNA transcript sequences predicted to be involved in defense of plant under the HS; 121 plant mRNA transcripts were found to be involved in 16 unique KEGG pathways. The annotation of the potential targets of miRNAs with differential expression indicates that genes encoding transcription factors, enzymes, and signal transduction components are implicated in the abiotic stress response. Some of the targets identified and implicated in thermotolerance pathways are small heat shock proteins (sHSPs like HSP17, HSP26), high molecular weight HSPs (HSP70, HSC, and HSP90), stress inducible protein, translocation protein, dnaj domain protein, betaine aldehyde dehydrogenase, etc. Interestingly, most of the identified mature miRNAs have small heat shock proteins, HSP70 and HSP90 genes, as their specific target which shows the importance of these chaperones in defense mechanism against HS in wheat. The known mature miRNA sequences identified in the control and HS-treated samples were aligned on 121 plant mRNA transcripts, and final results have been depicted in the electronic supplementary file as ESM_13.xls.
Comparative expression analysis of miRNAs and their target genes under the heat stress
Eight conserved miRNAs (tae-miR156, tae-miR160, tae-miR159a, tae-miR167, tae-miR1117, tae-miR164, tae-miR319, and tae-miR398) were randomly selected for the comparative expression analysis with their respective targets (HSP90, HSP70, WRKY, Dnaj, CPK1, HSP17, MYB3, and Cu/Zn-SOD) under the HS (Fig. 12). Under HS, the relative expression of tae-miR156 was high (2.2-fold) compared to their target (HSP90—1.38-fold). Similarly, tae-miR160 showed downregulation, and their target (HSP70) showed upregulation in response to HS. Similar pattern of expression was observed in tae-miR159a (target WRKY TF), tae-miR1117 (target CPK1), tae-miR164 (target HSP17), tae-miR319 (target MYB3) and tae-miR398 (target Cu/Zn-SODs). Maximum downregulation in response to HS was observed in tae-miR319 as compared to other miRNAs, whereas HSP17 showed maximum increase in the fold expression compared to other targets. We observed 3-fold increase in the expression of calcium-dependent protein kinase 1 (CPK1) which acts as signaling molecule in sensing the HS, and the miRNA regulating the expression of CPK1 (tae-miR1117) showed 0.5-fold downregulation under HS. Similarly, tae-miR164 showed downregulation (0.28-fold) compared to 11.1-fold increase in the expression of target gene (HSP17) under HS. In case of tae-miR167, we observed significant increase in the expression of miRNA and its target gene; relative expression was more in tae-mi167 compared to Dnaj (target) under HS. Similarly, Cu/ZN-SOD showed upregulation in response to HS, whereas the regulating miRNA (tae-miR398) showed downregulation under HS. Most of the miRNAs selected in the present investigation showed downregulation in response to HS, whereas there target genes showed upregulation under HS.

Quantitative real-time expression profiling of identified miRNAs and their target genes in seedlings of HD2985 cultivar of wheat exposed to heat stress (42 °C, 2 h); tae-miR156 (heat shock protein 90; accession no. DQ665783), tae-miR160 (heat shock protein 70; accession no. KJ027551), tae-miR159a (WRKY transcription factor; accession no. DQ286566), tae-miR167 (dnaj heat shock n-terminal domain-containing protein; accession no. KJ018078), tae-miR1117 (Calcium dependent protein kinase 1; accession no. JX878360), tae-miR164 (small heat shock proteins 17; accession no. JN572711), tae-miR319 (MYB3; accession no. AY615200), tae-miR398 (Cu/Zn-SOD; accession no. JQ613154). The expression of U6 SnRNA (for miRNA; Clontech, USA) and actin gene (for targets; accession no. AB181991) was used as endogenous control for normalizing the data. Relative expression was calculated by Pfaffl method
Quantitative real-time PCR validation of RNA-Seq results
Eight miRNAs were randomly selected for the comparative analysis of the differential expression values determined by RNA-Seq and qRT-PCR. The log2 fold change (FC) value of tae-miR156, tae-miR167, tae-miR398, tae-miR159a, tae-miR64, tae-miR319, tae-miR1117, and tae-miR160 observed by RNA-Seq and qRT-PCR was compared by histogram and scatter plot diagram (Fig. 13). Digital fold expression of miRNAs observed by RNA-Seq in response to HS was significant (p < 0.05) except tae-miR164 which showed non-significant result. Similarly, in case of qRT-PCR, the log fold change of miRNAs in response to HS was highly significant. The differences between the expression values observed by the two techniques were on par. The differential expression values of miRNAs observed by RNA-Seq and qRT-PCR were used for scatter plot analysis. We observed strong positive correlation between the differential expression values of miRNAs observed by the RNA-Seq and qRT-PCR. Linear regression shows that the both data are in agreement with each other (Fig. 13).

Quantitative real-time PCR validation of RNA-Seq results. (a) Comparison of differential expression values of miRNAs determined by RNA-Seq (dark gray) and qRT-PCR (white). Eight miRNA identified from Triticum aestivum were randomly selected for the analysis. (b) Scatter plot diagram showing the relationship between the log2 fold change (FC) of miRNA observed from RNA-Seq and qRT-PCR. Results of ANOVA are shown (**P < 0.05; *P > 0.05). Vertical bars indicate s.e (n = 3)
Discussion
Among different abiotic stresses, HS is considered as well as predicted the most detrimental factor reducing the yield of different crops, both in terms of quantity and quality (Lu et al. 2008). Plants, being sessile, have, however, developed biochemical and molecular mechanisms to cope up with such vagaries of nature (Coraggio and Tuberosa 2004). Overexpression of SAGs and stress-associated proteins (SAPs) has already been correlated with the thermotolerance mechanism of agriculturally important crops like wheat, rice, etc. (Kumar et al. 2014). Wheat, being thermosensitive, is severely affected especially when the temperature variations are more during pollination and grain-filling stages. The expression of SAGs has been established to be regulated by miRNAs. The number of miRNAs identified from wheat and deposited in repository is limited compared with other closely related species. In the present investigation, based on NGS data, we have identified 44 precursor and mature miRNAs in T. aestivum from miRBase v 19, 132 miRNA by homology-based approach using the reference genome of closely related species and 37 novel miRNAs using different analyzers and other plant-specific parameters. Deep sequencing method has been adequately exploited to study the conserved and novel miRNA in Arabidopsis, rice, Populus and Physcometrella, as is evident from the miRBase depository (Dryanova et al. 2008). To date, 5940 mature miRNA sequences have been identified from plant species. In this study, using bioinformatics tools, we have provided evidence for the existence of 33, 15, 11, and 41 % conserved miRNAs for O. sativa, Z. mays, S. bicolor, and A. thaliana, respectively. When compared with other plant species, tae-miR156b in wheat and osa-miR169 in rice were the most frequently observed miRNAs while miR156 exhibited low abundance in wheat as well as rice. Pandey et al. (2013) have reported five abiotic stress-responsive new miRNAs from wheat (Ta-miR5653, Ta-miR855, Ta-miR819k, Ta-miR3708, and Ta-miR5156). Ta-miR159 overexpressing rice lines were more sensitive to the HS, relative to the wild type, indicating that the downregulation of Ta-miR159 in wheat after HS might participate in a HS-related signaling pathway contributing, in turn, to the HS tolerance (Wang et al. 2012) which is in conformity with the observations in the present investigation. Kurtoglu et al. (2014) identified 52 putative wheat microRNAs using NGS, including seven, which have not been previously published. Zhao et al. (2013) identified 32 miRNAs from wheat and reported that 9 Ta-MIRs responded to Pi starvation: Ta-MIR159b, Ta-MIR167, Ta-MIR399, Ta-MIR408, Ta-MIR1122, Ta-MIR1125, Ta-MIR1135, Ta-MIR1136, and Ta-MIR1136 were upregulated, whereas Ta-MIR408 was downregulated. Xin et al. (2010) have also identified a set of miRNAs responsive to powdery mildew and HS in wheat. Sun et al. (2010) have provided the evidence for the existence of 20 conserved miRNA families as well as 23 novel miRNA families in wheat. Recently, they reported 323 wheat novel miRNAs and 524 target genes for 124 miRNA families by conducting a genome-wide survey of wheat miRNAs from 11 tissues (Sun et al. 2014).
We observed increase in the expression of target genes induced by the respective miRNAs under the HS. MiRNAs have been reported to be master regulator of plant growth and development (Sunkar et al. 2012), though it constitutes only 1 % of the total protein-coding genes of an organism (Shukla et al. 2008; Lukasik et al. 2013). The expression profiles of most of the miRNAs involved in plant growth and development were found to be altered in response to the HS. Wide variations in the transcripts of miRNAs and their respective targets were observed under the HS in the present investigation. Real-time PCR expression profiling showed increase in the transcript of signaling molecule like CDPK, whereas in case of HSPs, we observe decrease in the expression after 2 h of HS. The expression of some of the stress-responsive miRNAs has been reported to be regulated by calcium level in the cytosol (Mutum et al. 2013); this makes us to conclude that elevated temperature has marked effect on the expression of miRNAs which, in turn, trigger the expression of target genes associated with thermotolerance pathways. We found that a large, diverse, and complex small RNA population exists in T. aestivum. The observation that some of the miRNAs are upregulated or downregulated in response to the HS implies that these miRNAs are playing important roles in the stress tolerance.
Based on the reference genome sequence of rice, maize, and sorghum, we were able to identify 25 novel miRNAs from the control and 12 miRNAs from the HS-treated samples of HD2985 cultivar of wheat adding it to 37 novel candidate miRNAs. The present investigation has been able to establish the existence of novel heat-responsive miRNAs (candidate_430, candidate_3466, candidate_5064, candidate_5652, candidate_3182, and candidate_6941) in both thermotolerant and thermosusceptible cultivars of T. aestivum. Significant tissue-specific variations in the expression level of the identified miRNAs were observed in root, stem, and leaves of wheat seedling exposed to HS. Altered expressions of miRNAs during the stress have been implicated to have drastic effects on plant growth and development (Ding et al. 2013). It is well established that miRNAs are involved in plant response to the environmental stresses such as drought, salinity, heat, cold, nutrient starvation, oxidative stress, mechanical stress, UV-B radiation, etc. and trigger the expression of target genes involved in various tolerance mechanisms as observed in the present study (Sunkar and Zhu 2001; Lu et al. 2005; Lu et al. 2011; Pandey et al. 2013; Kumar et al. 2013a). We observed simultaneous decrease in the transcript of miRNA and increase in the expression of their targets under HS (Fig. 12) which is in conformity with the observation of Zhang et al. (2012). According to miRNA target databases, one miRNA may regulate many genes as its targets, while one gene may be targeted by many miRNAs. These findings indicate that relationships between miRNAs and their targets may not be one-to-one. Detailed comparison of the expression patterns of miRNAs and corresponding target genes revealed both negative and positive correlation between them (Zhang et al. 2012). Lopez-Gomollon et al. (2012) observed a negative correlation between the expression of miRNAs and their targets and reported that the correlation between the expression pattern of miRNA and their targets can vary between mRNAs belonging to the same gene family and even for the same target mRNA at different developmental stages. The expression of HSP17, CPK1, and HSP70 were observed high under HS which shows the potential role of these genes in thermotolerance. The expression of tae-miR167, tae-miR156, and their respective target genes (Dnaj and HSP90) showed upregulation under HS. The change of correlation during development suggests that the type of regulatory circuit directed by miRNA can change over time and can be different for individual gene family members (Lopez-Gomollon et al. 2012).
Guan et al. (2013) observed that expression of miR398 is quickly induced by HS at 37 °C and reaches its peak level 2 h after HS which is similar to our observation made in the present investigation. Although multiple abiotic stresses have been reported to upregulate the miR398 in Arabidopsis, Populus tremula, and Medicago truncatula plants (Yamasaki et al. 2007; Dugas and Bartel 2008; Jia et al. 2009; Trindade et al. 2010). Stief et al. (2014) reported that miR156 isoforms are highly induced after HS which confirms our observation. These heat-responsive miRNAs are predicted to be involved in regulating the expression of families of SAGs involved in modulating the defense mechanism of wheat under the HS.
Functional analysis has demonstrated that several plant miRNAs play vital roles in plant resistance to abiotic as well as biotic stresses (Shaik and Ramakrishna 2013). A number of heat-responsive transcripts have been demonstrated in TAM107 treated at 40 °C for 1 h, using Gene Chip Wheat Genome Array (Qin et al. 2008). The expression of heat-responsive miRNAs is induced by elevated temperature which, in turn, triggers the expression of their target genes and TFs. It is the abundance of these SAPs which decides the thermotolerance capacity of the plant under the HS. To sum up, in this paper, using de novo assembly and homology-based approaches, we have identified more than 132 mature miRNAs and 37 novel miRNAs in T. aestivum and validated six of the identified novel miRNA as heat-responsive. This will add to the available wheat miRNA data set. We also predicted targets of the respective miRNAs, and expression analysis showed abundance of sHSPs and signaling molecules under HS in wheat. We could also establish an increase in the expression of targets with downregulation of their respective miRNAs under the HS. Most of the targets identified appeared to involve in growth, development, metabolism, and defense-related processes under the abiotic stresses. Gaining insights into miRNA target genes can shed light on the range of miRNA regulation and may lead to a detailed description of miRNA-target interactions. We observed strong positive correlation between the differential expression values of miRNAs observed by the RNA-Seq and qRT-PCR. Expression analysis of newly identified miRNAs showed their differential regulatory role in different tissues under HS condition which might be involved in regulating metabolic and defense-related pathways of wheat.
Abbreviations
- NGS:
-
Next-generation sequencing
- RISC:
-
RNA-induced silencing complex
- miRNA:
-
MicroRNA
- HS:
-
Heat stress
- HSF:
-
Heat shock factor
- HSP:
-
Heat shock protein
- CDPK:
-
Calcium-dependent protein kinase
- qRT-PCR:
-
Quantitative real-time PCR
- SAGs:
-
Stress-associated genes
- SAPs:
-
Stress-associated proteins
References
Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC (2004) Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet 36:1282–1292
Barakat A, Wall PK, Diloreto S, Depamphilis CW, Carlson JE (2007) Conservation and divergence of microRNAs in Populus. BMC Genomics 8:481. doi:10.1186/1471-2164-8-481
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Berezikov E, Cuppen E, Plasterk RH (2006) Approaches to microRNA discovery. Nat Genet 38:S2–S7. doi:10.1038/ng1794
Carra A, Mica E, Gambino G, Pindo M, Moser C, Enrico MP, Schubert A (2009) Cloning and characterization of small non-coding RNAs from grape. Plant J 59:750–763
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Sci 301:336–338
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655
Coraggio I, Tuberosa R (2004) Molecular basis of plant adaptation to abiotic stress and approaches to enhance tolerance to hostile environments. Handb Plant Biotechnol. doi:10.1002/0470869143.kc022
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159. doi:10.1093/nar/gkr319
Ding Y, Tao Y, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64(11):3077–3086
Dryanova A, Zakharov A, Gulick PJ (2008) Data mining for miRNAs and their targets in the Triticeae. Genome 51(6):433–443
Dugas DV, Bartel B (2008) Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Mol Biol 67:1009–1014
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL et al (2007) High throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219
Gibson LR, Paulsen GM (1999) Yield components of wheat grown under high temperature stress during reproductive growth. Crop Sci 39(6):1841–1846
Guan Q, Lu X, Zeng H, Zhang Y, Zhu J (2013) Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J 74(5):840–851
Jia X, Wang WX, Ren L, Chen QJ, Mendu V, Willcut B, Dinkins R, Tang X, Tang G (2009) Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol Biol 71:51–59
Jia J, Zhao S, Kong X, Li Y, Zhao G et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209–216
Kidner CA, Martienssen RA (2005) The developmental role of microRNA in plants. Curr Opin Plant Biol 8:38–44
Klevebring D, Street NR, Fahlgren N, Kasschau KD, Carrington JC, Lundeberg J, Jansson S (2009) A genome-wide profiling of Populus small RNAs. BMC Genomics 10:620. doi:10.1186/1471-2164-10-620
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. NAR 39(Database Issue):D152–D157
Kumar RR, Goswami S, Sharma SK, Singh K, Gadpayle KA, Singh SD, Pathak H, Rai RD (2013a) Differential expression of heat shock protein and alteration in osmolyte accumulation under heat stress in wheat. J Plant Biochem Biotechnol 22:16–26
Kumar RR, Sharma SK, Goswami S, Singh GP, Singh R, Singh K, Pathak H, Rai RD (2013b) Characterization of differentially expressed stress associated proteins in starch granule development under heat stress in wheat (Triticum aestivum L.). Indian J Biochem Biophys 50:126–138
Kumar RR, Singh GP, Goswami S, Pathak H, Rai RD (2014) Proteome analysis of wheat (Triticum aestivum) for the identification of differentially expressed heat-responsive proteins. Aus J Crop Sci 8(6):973–986
Kurtoglu KY, Kantar M, Budak H (2014) New wheat microRNA using whole-genome sequence. Funct Integr Genomics 14(2):363–379
Lopez-Gomollon S, Mohorianu I, Szittya G, Moulton V, Dalmay T (2012) Diverse correlation patterns between microRNAs and their targets during tomato fruit development indicates different modes of microRNA actions. Planta 236(6):1875–1887. doi:10.1007/s00425-012-1734-7
Lu S, Sun YH, Shi R, Clark C, Li L et al (2005) Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 17:2186–2203
Lu S, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Plant J 55:131–151
Lu S, Yang C, Chiang VL (2011) Conservation and diversity of microRNA-associated copper-regulatory networks in Populus trichocarpa. J Integr Plant Biol 53:879–891
Lukasik A, Pietrykowska H, Paczek L, Szweykowska-Kulinska Z, Zielenkiewicz P (2013) High-throughput sequencing identification of novel and conserved miRNAs in the Brassica oleracea leaves. BMC Genomics 14(1):801. doi:10.1186/1471-2164-14-801
Mayer KF, Rogers J, Dolezel J, Pozniak C, Eversole K, Feuillet C et al (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Sci 345(6194):1251788. doi:10.1126/science.1251788
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman J, Cao X, Carrington JC, Chen X, Green PJ et al (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Mutum RD, Balyan SC, Kansal S, Agarwal P, Kumar S, Kumar M, Raghuvanshi S (2013) Evolution of variety specific regulatory schema for expression of osamiR408 in indica rice varieties under drought stress. FEBS J 280(7):1717–1730
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R et al (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Pandey B, Gupta O, Pandey D, Sharma I, Sharma P (2013) Identification of new stress-induced microRNA and their targets in wheat using computational approach. Plant Signaling Behav 8:e23932, PMID:23511197
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45–e45
Qin D, Wu H, Peng H, Yao Y, Ni Z, Li Z, Sun Q (2008) Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using wheat genome array. BMC Genomics 9(1):432. doi:10.1186/1471-2164-9-432
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Shaik R, Ramakrishna W (2013) Machine learning approaches distinguish multiple stress conditions using stress responsive genes and identify candidate genes for broad resistance in rice. Plant Physiol 164(1):481–495. doi:10.1104/pp.113
Shukla LI, Chinnusamy V, Sunkar R (2008) The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim Biophys Acta Gene Regul Mech 1779(11):743–748
Song C, Wang C, Zhang C, Korir N, Yu H, Ma Z, Fang J (2010) Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 11(1):431–443. doi:10.1186/1471-2164-11-431
Srivastava N, Chaudhary S, Kumar V, Katudia K, Vaidya K, Vyas MK, Chikara SK (2012) Evaluation of the yield, quality and integrity of total RNA extracted by four different extraction methods in rice (Oryza sativa). Res Rev J Crop Sci Technol 1:1–9
Stief A, Altmann S, Hoffmann K, Pant BD, Scheible WR, Baurle I (2014) Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell Online 26(4):1792–1807
Sun W, Julie Li Y-S, Huang HD, Shyy JYJ, Chien S (2010) microRNA: a master regulator of cellular processes for bioengineering systems. Ann Rev Biomed Eng 12:1–27
Sun F, Guo G, Du J, Guo W, Peng H, Ni Z, Sun Q, Yao Y (2014) Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol 14:142
Sunkar R, Zhu JK (2001) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17(4):196–203
Trindade I, Capitao C, Dalmay T, Fevereiro MP, Santos DM (2010) miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231:705–716
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wahid A, Gelani S, Ashraf M, Foolad MR (2007) Heat tolerance in plants: an overview. Env Exp Bot 61(3):199–223
Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW et al (2005) Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 17:2204–2216
Wang Y, Sun F, Cao H, Peng H, Ni Z et al (2012) TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS One 7(11):e48445. doi:10.1371/journal.pone.0048445
Xin M, Wang Y, Yao Y, Xie C, Peng H, Ni Z, Sun Q (2010) Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol 10:123–134
Yamasaki H, Abdel-Ghany SE, Cohu CM, Kobayashi Y, Shikanai T, Pilon M (2007) Regulation of copper homeostasis by micro-RNA in Arabidopsis. J Biol Chem 282:16369–16378
Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, Sun Q (2007) Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol 8:R96. doi:10.1186/gb-2007-8-6-r96
Zhai J, Zhao Y, Simon SA, Huang S, Petsch K, Arikit S et al (2013) Plant microRNAs display differential 3′ truncation and tailing modifications that are ARGONAUTE1 dependent and conserved across species. Plant Cell 25(7):2417–2428. doi:10.1105/tpc.113
Zhang JZ, Ai XY, Guo WW, Peng SA, Deng XX, Hu CG (2012) Identification of miRNAs and their target genes using deep sequencing and degradome analysis in trifoliate orange (Poncirus trifoliata L. Raf). Mol Biotechnol 51(1):44–57. doi:10.1007/s12033-011-9439-x
Zhao CZ, Xia H, Frazier TP, Yao YY, Bi YP et al (2010) Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant Biol 10:3–15. doi:10.1186/1471-2229-10-3
Zhao X, Liu X, Guo C, Gu J, Xiao K (2013) Identification and characterization of microRNAs from wheat (Triticum aestivum L.) under phosphorus deprivation. J Plant Biochem Biotechnol 22(1):113–123
Zuker M (1989) On finding all suboptimal folding of an RNA molecule. Sci 244(4900):48–52
Acknowledgments
Financial assistance provided by the Indian Council of Agriculture Research (ICAR) under the National Initiative for Climate Resilient Agriculture (NICRA) project (Project 12/115 TG3079) is highly acknowledged. We also thank Director, Indian Agricultural Research Institute (IARI) for providing all the logistic support required for executing the research work.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
Experiments comply with the current laws of the country in which they were performed.
Availability of NGS raw data
The data sets supporting the results of this article are available in the National Center for Biotechnology Information (NCBI) repository [BioProject Database: PRJNA172054; http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA172054].
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM_1.tif
Flow chart of the work plan undertaken for the identification of conserved and novel heat-responsive miRNA in control and heat stress treated samples of HD2985 (thermotolerant) cultivar of wheat (Triticum aestivum L.). (DOCX 158 kb)
ESM_2.tif
Work-flow for novel miRNA discovery using the raw reads from the sequenced libraries of control and heat stress treated samples of HD2985 cultivar of wheat (Triticum aestivum L.); different pipeline analyzers were used for the identification of novel heat-responsive miRNAs from the potential candidates. (DOCX 45 kb)
ESM_3.xls
List of primers used for the comparative expression analysis of mature miRNAs and their target genes in control and heat stress treated samples of HD2985 (thermotolerant) cultivar of wheat (Triticum aestivum). (XLS 39 kb)
ESM_4.xls
List of miRNAs identified in control and heat stress treated samples of HD2985 (thermotolerant) cultivar using the reference draft genome of wheat (Triticum aestivum). (XLS 44 kb)
ESM_5.xls
A list of candidate miRNAs, identified targets and their functional annotation based on the reference draft genome of Triticum aestivum (retrieved from ftp://ftp.ensemblgenomes.org). (XLS 50 kb)
ESM_6.xls
List of miRNAs identified in control and heat stress treated samples of HD2985 (thermotolerant) cultivar of wheat based on homology search using the reference genome of rice (Oryza sativa). (XLS 48 kb)
ESM_7.xls
List of miRNAs identified in control and heat stress treated samples of HD2985 (thermotolerant) cultivar of wheat based on homology search using the reference genome of maize (Zea mays). (XLS 42 kb)
ESM_8.xls
List of miRNAs identified in control and heat stress treated samples of HD2985 (thermotolerant) cultivar of wheat based on homology search using the reference genome of sorghum (Sorghum bicolor). (XLS 41 kb)
ESM_9.xls
List of target genes of mature miRNAs identified by homology based search using the reference genome of T. aestivum, O. sativa, Z. mays and S. bicolor. (XLS 229 kb)
ESM_10.tif
Secondary structure analysis of validated heat-responsive miRNAs from wheat (Triticum aestivum); Flicker v3.0 was used for the prediction of secondary structure and structure analysis was performed using Zuker’s algorithm. (DOCX 95 kb)
ESM_11.xls
Novel miRNAs (identified based on homology search using the reference genome of O. sativa, Z. mays and S. bicolor) and their respective target genes identified in control sample of HD2985 cultivar of wheat. (XLS 122 kb)
ESM_12.xls
Novel miRNAs (identified based on homology search using the reference genome of O. sativa, Z. mays and S. bicolor) and their target genes identified in heat shock treated samples of HD2985 cultivar of wheat. (XLS 60 kb)
ESM_13.xls
List of mature miRNAs (identified in present investigation) involved in thermotolerance pathways along with their target genes. (XLS 335 kb)
Rights and permissions
About this article

Cite this article
Kumar, R.R., Pathak, H., Sharma, S.K. et al. Novel and conserved heat-responsive microRNAs in wheat (Triticum aestivum L.). Funct Integr Genomics 15, 323–348 (2015). https://doi.org/10.1007/s10142-014-0421-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-014-0421-0