
Abstract
Grapevine is one of the economically and culturally important perennial fruit crops. More than 60 viruses infect grapevines and adversely affect their growth and development. Latent infection of most viruses in grapevines leads to chronic modulation of gene expression at transcriptional and post-transcriptional levels. Plant small RNAs (sRNAs) consist of microRNA (miRNA) and small interfering RNA (siRNA). miRNAs are expressed from the plant genome while most siRNAs are derived from double-stranded RNA molecules which are intermediates during virus replication. In a previous study, we constructed four cDNA libraries of sRNAs that were enriched from three virus-infected grapevines and one virus-free grapevine. Majority of siRNAs align most closely with the genomes of DNA viruses in the genus Badnavirus, family Caulimoviridae that led to the discovery of a new Grapevine vein clearing virus in grapevines. In this study, we conducted a comprehensive analysis of miRNAs in the four cDNA libraries and identified novel and stress-related miRNAs. The results indicated that miRNA abundance was influenced by virus infection. A total of 54 new miRNAs were identified and characterized, six of which, VITIS-MIR17, 18, 19, 20, 21, and 22, were detected only in virus-infected samples. One target of VITIS-MIR18 is the gene coding a non-apical meristem protein (GSVIVT00035370001), a transcription factor in the regulation of plant development and stress responses. Among the virus infection-induced known miRNAs, miRNA168 and miRNA3623 likely regulate grapevine’s defense response, miRNA319 and miRNA395 modulate the expression of genes that are involved in nutrient metabolisms while miRNA396 plays a role in the regulation of cell division and cell cycle. The abiotic stress-induced miR169 and mi398 were negatively regulated by virus infection in grapevines. In addition, variety-specific miRNAs were discovered and compiled. The newly discovered miRNAs expand the miRNA profiles in the Vitis species. The characteristics of variety-specific and virus infection-associated miRNAs help understand the biology underlying the development and defense response of grapevines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant small RNAs (sRNAs) in the range of 18–24 nucleotides (nt) are classified into two major groups: microRNAs (miRNAs) and small interfering RNAs (siRNAs), although the distinction between miRNAs and siRNAs becomes less important in their functionality and mode of operation (Carthew and Sontheimer 2009; Bartel 2009; Voinnet 2009). sRNAs play important roles in the developmental regulation and environmental adaptation of plants. miRNA exerts its regulation of protein expression by either guiding the RNA-induced silencing complexes (RISC) to complementary mRNA to degrade target mRNA or inhibit the translation of mRNA. miRNAs are products of the complementary stem portion of pri- and pre-miRNA molecules that have stem-loop hairpin structures where miRNA is the guide strand incorporated into RISC and miRNA* is the byproduct of miRNA biogenesis (Voinnet 2009). siRNAs can originate either from much longer hairpins that produce largely divers siRNAs, or from fully or near-perfect RNA duplexes (Bartel 2009).
miRNAs of 18–21 nt are processed mainly by Dicer-like (DCL) protein 1 or DCL4, whereas 22- and 24-nt miRNAs are products of DCL2 and 3 (Voinnet 2009). A group of miRNAs are ancient signature sRNAs that are highly conserved and abundant in the land plants (Rajagopalan et al. 2006; Fahlgren et al. 2007). In contrast, evolutionarily young miRNAs emerged as result of recent genomic rearrangement and are in low expression, have fewer targets than deeply conserved miRNA (Rajagopalan et al. 2006; Fahlgren et al. 2007, 2010). Furthermore, duplex forms of a subset of 21, 22, 24-nt miRNAs are mobile from cells to cells (Dunoyer et al. 2010), and along the plant vascular system (Molnar et al. 2010). The mobility of miRNAs exerts extensive effects on adaptation and the fate of cells beyond the site of miRNA biogenesis, which has significant implications in plant biology.
Virus infection alters the miRNAs profiles in plants (Bazzini et al. 2007; Tagami et al. 2007). Infection of tobacco (Nicotiana tobacum) plants with Tobacco mosaic virus (TMV), Tomato mosaic virus, Tobacco etch virus, Potato virus Y, and Potato virus X changed the abundance of 10 conserved miRNAs to various degrees (Bazzini et al. 2007). Levels of certain miRNAs increased significantly in TMV-infected Arabidopsis and four novel miRNAs were also identified as a result of TMV infection (Tagami et al. 2007). Thus, it was proposed that virus-infected plants are good sources for identifying new miRNAs. The infection of Arabidopsis with TMV or Oilseed rape mosaic virus (ORMV) activated the promoter of miRNA164a and influences the transcription of miRNAs (Bazzini et al. 2009). On the other hand, endogenous miRNAs, if complementary with regions of viral genomes, could guide RISC to target plant virus genomes in plant’s innate defense against viruses (Perez-Quintero et al. 2010).
Genome sequencing of Vitis vinifera grapevine “ENTAV115” and “PN40024” unveiled the presence of four DCLs, nine Argonaute (AGO), six RNA-dependent RNA polymerase genes (Jaillon et al. 2007; Velasco et al. 2007) indicating complex RNA processing system is present in grapevines. To date, a total of 163 miRNAs have been compiled from the ENTAV115 sequencing project (Velasco et al. 2007) and from the studies on the identification of grapevine miRNAs (Carra et al. 2009; Pantaleo et al. 2010; Mica et al. 2010) that have been deposited in the Vitis repository at miRBase. More novel miRNAs have been discovered in an inter-specific hybrid table grape “Summer Black” in a recent study (Wang et al. 2011a, b). Most grapevine miRNAs were predicted to target mRNAs similar to those in Arabidopsis (Velasco et al. 2007). Tissue-specific expressions of grapevine miRNAs exemplified the involvement of miRNAs in the processes of inflorescence and tendril development (Pantaleo et al. 2010), as well as leaf and berry development (Carra et al. 2009; Pantaleo et al. 2010; Mica et al. 2010; Wang et al. 2011a, b). Grapevine miRNAs were also associated with the defense responses against pathogens (Carra et al. 2009).
Grapevine, a perennial fruit plant, faces various abiotic and biotic stresses along decades-long lifespan. Grapevine has developed strategies of coping with and adapting to these stresses. It is apparent that environmental cues and pathogen attacks induce different sets of miRNAs in grapevine. The heterozygocity of the grape genome exerts additional effects on the spectrum of miRNAs, as evidenced by the recent finding that the 21-nt miRNAs are more abundant than 24-nt miRNAs in grapevine ENTAV115 (Pantaleo et al. 2010). As addressed by Voinnet (2009), the identification of novel miRNAs and the potential new modes of action in the regulation of biological activities require utilization of crop plants that are grown under field conditions that experience bona fide environmental stresses and face challenges by various pathogens. These facts make grapevine a model fruit crop that entails deep sequencing of more grape varieties that are under different stresses to enrich the plant miRNA families for unveiling miRNA-mediated regulation of development and adaption that are normally eluded by model plants grown under the ideal laboratory conditions.
We have described the analysis of sRNAs and discovery of new viruses in grapevines in a companion publication (Zhang et al. 2011a). In this article, we describe the results from the analysis of miRNAs in the sRNA libraries that were constructed from three individual virus-infected grapevines and from one virus-free grapevine after the high-throughput sequencing via the Illumina Solexa platform. We identified 54 new grapevine miRNAs along with miRNA* sequences. The 21-nt miRNAs represent the most abundant classes among the miRNA population in each sRNA library. A set of conserved and grapevine-specific miRNAs were compiled and found to be associated with virus infection. Their potential targets are related to the basal plant defense and the responsiveness to nutrient deficiency. Some miRNAs are found only in a particular variety, indicating their variety-specific expression. Discovery of virus infection-associated miRNAs in grapevines provided insights on molecular interaction between grapevines and viruses.
Materials and methods
Grapevines
Four grapevines VFM411, LBC0903, SHC0903, and SHV0904 were selected for this study. VFM411, LBC0903, and SHC0903 are variety “Chardonel”, a hybrid of SeyvalT and V. vinifera “Chardonnay”. SHV0904 is a Vidal Blanc vine, a French hybrid of Ugni Blanc (also Trebbiano), an ancient grape variety originated in the Eastern Mediterranean, and Rayon d’Or (also Seibel 4986). VFM411 was propagated from a 0.5 mm microshoot tip of a Chardonel vine after the vine was treated under high temperature (37 °C/16 h, light and 22 °C/8 h, dark) for 1 month. VFM411 is free of most grapevine viruses as evidenced by the analysis of siRNA profiles in the high throughput sequencing (Zhang et al. 2011a, b), and thus is referred to as the virus-free grapevine. Grapevines LBC0903, SHC0903, and SHV0904 were propagated from green cuttings that were collected from the symptomatic vine that was grown in two commercial vineyards at RochPort and Herman, MO, USA and all show virus-like symptoms, most notably are short zig-zag internodes on shoots and translucent vein clearing on young leaves (Fig. 1). These were grown in the potted soils under the same conditions in greenhouses. Grapevine VFM411, originally propagated from the microshoot tip, was also grown in potted soils in the same greenhouse. The third or fourth leaf on the young shoots was collected and immediately frozen in liquid nitrogen and stored at −80 C.

Appearance of translucent vein clearing symptom on the grapevine LBC0903, SHC0903, and SHV0904 in three commercial vineyards
RNA extraction, cDNA library construction and solexa sequencing
Total RNA was extracted from the stored leaves as described previously (Fung et al. 2008). Detailed description of procedures and protocols for enriching small RNA, constructing cDNA libraries, and the Solexa sequencing of cDNA libraries were documented in a previous publication (Zhang et al. 2011a, b).
Processing of raw sequence and identification of miRNAs
The raw Illumina files of sRNA were retrieved from the GNomex site at Microarray core facility, Huntsman Cancer Institute, University of Utah, Saltlake City, USA. For the analysis of the sRNA data, the standalone and online version of the UEA sRNA Toolkit was used, which is a package of several tools for the analysis of high-throughput small RNA data. The adaptor sequences were removed by finding exact matches of the 3′ adaptor and optionally the 5′ adaptor sequences on the raw sequences. sRNAs in the range of 16 to 28 nt were extracted and the sRNA abundance was calculated.
The miRNA database, miRBase release 17 contains 16,772 entries of hairpin precursor miRNAs in 153 species and over 19,000 mature miRNA sequences. This database has been used to search for known and conserved miRNAs. miRBase has 163 unique entries for V. vinifera miRNAs.
The expression profiles of sRNAs that match known miRNAs in miRBase database were produced by the miRProf tool. The 12× V. vinifera PN40024 genome sequences (Genoscope 12×, v12_02_10) were used for database search and annotation with two mismatches allowed. The sRNAs were filtered to retain the valid sequences where the sequences longer than 50 nt and low complexity sequences were removed. The tRNA/rRNA sequences were removed by searching against known plant tRNA and rRNA sequences and filtering out the sequences that do not match the genome selected. The miRNAs were generated and the total read count after the final filtering step was used for normalized counts which were given in matching reads per one million total reads.
Prediction and annotation of target genes
The miRProf miRNA sequences in the UEA sRNA Tookit were used to predict their respective targets using V. vinifera as the Transcript database. Importantly, miRNA and target duplexes which have no more than four mismatches between them and with no more than two adjacent mismatches in the duplex are considered.
The Grape Genome Browser, Genoscope is used to perform annotation of the generated targets by BLAT search against the Vitis transcript database. BLAT is designed to quickly find sequences of 95 % and greater similarity of length 40 bases or more by keeping an index of the entire genome in memory. The index consists of all non-overlapping 11-mers. The target query which has the highest score and alignment identity was chosen to identify its Vitis transcript match. The identified transcript was used for annotation with PFAM, PROSITE, and SMART. The annotation results were also verified with VitisNet-Molecular Networks for Grapevine.
Prediction of new miRNA
The new miRNAs were predicted by the miRCat toolkit based on abundance and secondary structure (Moxon et al. 2008). The miRNAs that were not present in the miRBase database or had a very low identity with the known miRNAs were considered to be new/predicted miRNAs. The miRNAs that had high abundance (number of times sequenced in high-throughput sequencing), low minimum free energy of the predicted hairpin sequence, and had their corresponding miRNA* sequence(s) were deemed as potential new miRNAs.
Results and discussion
Deep sequencing of sRNAs from one virus-free and three virus-infected grapevines
We extracted total RNA enriched with sRNAs from young leaves of the four grapevines, VFM411, LBC0903, SHC0903, and SHV0904. A cDNA library of sRNAs was constructed for each grapevine. The Solexa sequencing of sRNA populations showed that these samples contain abundant amounts of siRNAs that match with genomes of multiple viruses (Zhang et al. 2011a). Among them, a group of viruses in the family Caulimoviridae was the most abundant, and a newly isolated DNA virus, Grapevine vein clearing virus was confirmed to be present in the grapevine LBC0903, SHC0903, and SHV0904 (Zhang et al. 2011a, b).
High-throughput sequencing of sRNA populations yielded more than five million raw reads for cDNA samples of LBC0903, SHC0903, SHV0904, and less than one million for VFM411 (Table 1). Those reads having 3′ adapter sequences and in the size range of 16–28 nt were selected for further analysis. Upon removing the reads matching to known plant tRNA and rRNA sequences, we discovered a total of 1,483,611, 1,642,680, 2,297,101, and 380,714 of redundant sRNAs from the leaf tissue of LBC0903, SHC0903, SHV0904, and VFM411, respectively, that were mapped to the 12× PN40024 reference genome (Jaillon et al. 2007). Percentage of nonredundant sRNAs was calculated to be in the range of 22–33 % of total sRNAs.
The 21-nt class of sRNAs is the most abundant
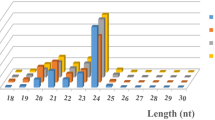
From the total number of sRNAs in each sample, the number of each class of sRNA was calculated and presented in Fig. 2. The 21-nt sRNAs are the most abundant among the three virus-infected samples LBC0903, SHC0903, and SHV0904. These results are in agreement with the previous findings that the 21-nt sRNAs were the most prominent class among grapevine sRNAs (Pantaleo et al. 2010; Wang et al. 2011a, b). Nearly equal number of 22- and 24-nt sRNAs is found in three virus-infected grapevines. Interestingly, the number of 24-nt sRNAs is the highest in the virus-free sample VFM411 cDNA library (Fig. 2b). We found that the total numbers of sRNA in the virus-infected grapevine leaves that match with the 12× grapevine reference genome PN40024 are remarkably higher than in the virus-free grapevine VFM411 (Table 1). One explanation for the higher number of 24-nt in VFM411 is that the total number of sRNA may skew the distribution of 21-nt sRNAs and 24-nt sRNAs. It is also possible that virus infection influences the ratio of 21 and 24-nt sRNA classes.

Size distribution of redundant small RNAs. a Comparison of four grapevines LBC0903, SHC0903, SHV0904, and VFM411. b Sample VFM411 with a higher number of 24-nt sRNAs than 21-nt sRNAs. Y-axis is the number of sRNA reads for each sRNA class, X-axis is the size of sRNAs in nucleotides
Profiles of novel miRNAs
From sRNA sequences of the four cDNA libraries that were mapped to the 12× PN40024 genome, we found 50 miRNAs that were previously documented in other plants and grapevine. Based on the primary criteria for annotating miRNAs that miRNAs shall form a stem-loop precursor with miRNA* (Meyers et al. 2008), we discovered a total of 54 novel miRNAs that can form predicted secondary structures with their corresponding miRNA* (Electronic supplementary material (ESM) Table 1). These new miRNAs were given provisional VITIS-MIR number from 1 to 54. All new miRNA were mapped to a single locus on the PN40024 reference genome except for VITIS-MIR11, which is mapped to five genomic loci on Chromosome 19. Among the 54 new miRNAs, eight are specific in LBC0903, 14 in SHC0903, six in SHV0904, and two in VFM411. Fifteen out of 54 new miRNAs were predicted to target grapevine genes (ESM Table 2).
The locus for VITIS-MIR1 and VITIS-MIR34 (in red text) is only three base pairs apart on chromosome 10 while the locus for VITIS-MIR3 is 86 bp upstream of the locus for VITIS-MIR 37 on chromosome 14. VITIS-MIR5 is mapped to chromosome 16 at position 5413775–5413798 while VITIS-MIR38 to Chr16 at 5413739–5413333761, only 14 base pairs apart, at same direction. Similarly, VITIS-MIR6 and VITIS-MIR11-5 are located only 48 pb apart on chromosome 19. VITIS-MIR7 and VITIS-MIR44 are 28 bp apart on chromosome 4. These findings are similar to a previous report that two new miRC13 and miRC15 are located closely on chromosome 17 (Pantaleo et al. 2010), raising the question if these closely clustered miRNAs are generated from the same miRNA precursor. Interestingly,VITIS-MIR1, 3, 5, 6, and 7 are found only in the sample LBC0903 while VITIS-MIR34, 37, 38, 11-5, and 44 are in the sample SHC0903 (ESM Table 2), suggesting that they are expressed differentially in individual vines, either from their own promoter or simply activated in different vines, even though they are closely located on the same chromosome.
As of July 19, 2011, there are 163 miRNA of Vitis species in the miRBase (http://www.mirbase.org/cgi-bin/mirna_summary.pl?org=vvi). The 163 miRNAs are compiled from the grapevine PN40024 or Pinot Noir clone ENTAV115. Both are derived from V. vinifera, which is the Eurasian Vitis species. In a recent study, Wang et al. (2011a) discovered 80 miRNAs with most of the potential novel miRNAs in a table grape variety “Summer Black”, a hybrid of V. vinifera and V. labrusca. In this study, we identified 54 potential new miRNAs (ESM Tables 1 and 2). It is known that compositions and expression patterns of miRNAs are influenced largely by abiotic and biotic factors (Sunkar and Zhu 2004; Sunkar et al. 2007; Katiyar-Agarwal and Jin 2010). Leaf tissues of Chardonel and Vidal Blanc, two interspecific hybrids of V. vinifera with other Vitis species, including North American Vitis species, were sampled for the Solexa sequencing of sRNA. Several miRNAs were discovered only in Chardonel variety while others in Vidal Blanc, suggesting that the heterozygocity of grapevine genomes derived from different Vitis species significantly affect the composition and abundance of miRNAs. Furthermore, the profile of virus-associated miRNAs reflects the influence of virus infection on the expression of miRNA that also leads to the finding of new miRNAs. Voinnet (2009) suggested that characterization of sRNAs in crop plants grown in the field can identify new miRNAs and discover new mode of regulation of miRNAs. The discovery of 54 new miRNA from the present study corroborate the point that heterogenetic background, pathogen infection, and environmental conditions shape the miRNA populations.
New VITIS-MIR55 has exceptionally high number of reads (8,784 reads; ESM Table 2), its target gene encodes a zinc finger transcription factor that is known to be critical in basal transcription. The high expression of miRNA45 in grapevine makes it a good candidate for further study of its biological roles in grapevine growth and development.
Virus infection-induced miRNAs and their target genes
Because the three selected grapevine samples LBC0903, SHC0903, and SHV0904 were infected by viruses (Fig. 1), we reasoned that insightful information can be drawn from the prediction of the miRNA profiles that are associated with virus infection. We considered a miRNA to be upregulated by virus infection if the normalized count of the miRNA is twice more in a virus-infected grapevine than in the virus-free VFM411. We discovered a total of 23 miRNA that were expressed at a higher level in at least one of three virus-infected grapevines than in the virus-free VFM411 sample (Table 2). Among the virus-associated miRNAs, the most notable are miRNA319, miRNA396, and miRNA3623 that have a fold change of 10 or more in all three virus-infected grapevines. Interestingly, no reads of miRNA395, miRNA1511/miRNA5139, and miRNA3624 were found in the VFM411, and they were all detected in the virus-infected grapevines. In addition, we found that new VITIS-MIR17, 18, 19, 20, 21 and 22 were detected only in virus-infected samples LBC0903, SHC0903, and SHV0904 (ESM Table 2).
Our data showed that miRNA168 is upregulated in the virus-infected grapevines (Table 2). miRNA168 is one of miRNAs that are closely associated with virus infection. It was found that miRNA168 was induced significantly in Nicotiana benthamiana plants after they were infected with TMV, Potato virus X (PVX) and Tobacco etch virus (TEV), and also in Arabidopsis plants infected with Ribgrass mosaic virus, and Turnip crinkle virus, in Medicago tuncatula with TMV, and in Solanum lycopersicum with PVX (Varallyay et al. 2010). Expression level of miRNA168 also was elevated in N. benthamiana infected by CbLCuV and TYLCV (Amin et al. 2011), in Arabidopsis plants with ORMV (Hu et al. 2011), and in tomato plants with Cucumber mosaic virus satellite RNAs (Feng et al. 2012). Therefore, the induction of miRNA168 is a ubiquitous phenomenon in plant–virus interaction, suggesting that miRNA168 plays an import role in the host defense and virus’s counter defense (Varallyay et al. 2010). It is hypothesized that the virus-induced accumulation of miRNA168 may repress the translation of Ago1 mRNA and thus inhibits the expression of AGO1 protein. Since AGO1 recruits virus-derived siRNA and then degrades the target viral RNA, a central node of the RNA-silencing in host defense against virus (Zhang et al. 2006), the general induction of miRNA168 in virus-infected plants may counter the inhibitory activity by AGO1 on the virus replication (Varallyay et al. 2010). The high abundance of miRNA168 in the virus-infected grapevines is in agreement with the previous findings on the virus-induced miRNA168 expression.
miRNA319, miRNA396, and miRNA3623 are the most noticeable among the upregulated miRNAs in virus-infected grapevines (Table 2). A predicted target of miRNA319 is nucleobase-ascorbate transporter (NAT) family (Table 4). The NAT family proteins are ubiquitous plasma transmembrane proteins for uptaking uracil, xanthine, or uric acid in plant, and thus play crucial roles in the intercellular traffic of nutrients (Gournas et al. 2008). miRNA319 was upregulated in tomato with the infection of Tomato leaf curl New Delhi virus, a member of Geminiviruses (Naqvi et al. 2010), and also in virus-infected grapevines in this study. It can be speculated that downregulation of NAT family proteins as a result of increased expression of miRNA319 may have adverse effect on the regular transmembrane transferring of nutrients, which is part of the virus pathogenesis. miRNA396 targets Arabidopsis growth-regulating factors and negatively regulates cell division and cell cycle (Wang et al. 2011b). A target gene of miRNA3623 encodes a TIR-NBS-LLR protein (ESM Table 3), which is well characterized in the plant’s defense against pathogens.
Interestingly, miR395, miR1511, and miR3624 were detected only in the virus-infected LBC0903, SHC0903, and SHV0904, suggesting their close association with virus infection. One of the predicted target genes of miRNA395 is sulfate adenylyltransferase 3 in the sulfate metabolism pathway (ESM Table 3). In Arabidopsis, miRNA395 targets the ATP sulfurylase genes APS1, APS3, and APS4, and sulfate transporter 2;1 (SULTR2;1) genes, and downregulates the transcripts of APS and SULTR2;1 genes, and thus regulates the sulfate accumulation and relocation (Liang et al. 2010; Kawashima et al. 2011). Furthermore, miRNA395 is enriched in the phloem and can move from scion to rootstock through the graft union and thus likely a phloem-mobile signaling molecule in the plant’s response to nutrient deprivation (Buhtz et al. 2010).
In addition, we found that new VITIS-MIR17 to 22 were detected only in virus-infected samples LBC0903, SHC0903, and SHV0904 (ESM Tables 1 and 2). One target of the virus-associated VITIS-MIR18 is the gene coding a non-apical meristem (NAM) protein (GSVIVT00035370001). NAM proteins are plant specific transcription factors that play crucial roles in the regulation of plant development and responses to abiotic and biotic factors (Olsen et al. 2005). It was shown that Arabidopsis ortholog of this virus infection-associated grapevine NAM is a transcriptional regulator in the circuit of pathway for responding to cold stress and regulating flowering time (Yoo et al. 2007). Thus, the functionality of the targeted gene makes the new VITIS-MIR18 a good candidate for further investigation of its role in viral pathogenesis.
Virus infection-suppressed miRNAs and their target genes
At least 10 miRNAs were downregulated by virus infection as they were detected only, or at high abundance, in the virus-free sample VFM411 (Table 3 and 4). miR169 negatively regulates stomatal opening during drought response in tomato and thus potentially improves drought tolerance (Zhang et al. 2011b). miRNA169 is also involved in the nitrogen metabolism in Arabidopsis (Zhao et al. 2011). miRNA398 is one of the most studied stress-responsive miRNA in plants, it is speculated to be associated with regulation of plant stress responses to abiotic stresses such as copper and phosphate and biotic stress bacterial infection (Zhu et al. 2011). Furthermore, both miRNA398 and miRNA408 are up-regulated under water deprivation conditions in Medicago truncatula (Trindade et al. 2010).
miRNA could also be involved directly in the antiviral defense response in plants. A bioinformatics analysis of miRNA in over 30 plant species including V. vinifera and search for their perfect match with regions on viral genomes suggested that plant miRNA may target plant viral genome directly with similar function to the virus-derived siRNA in guiding RISC to the targeted viral genome (Perez-Quintero et al. 2010). Based on the bioinformatics analysis, at least seven grapevine viruses, Grapevine leaf roll-associated virus 3, Grapevine rootstock stem lesion associated virus, GLRaV-2, GLRaV-10, Grapevine fleck virus, Grapevine virus A, Arabis mosaic virus are potential targets of miRNAs. These viruses were detected in all the sRNA libraries we prepared (Kashmir, unpublished results). For instance, the highly conserved miRNA395, which is detected only in the virus-infected grapevines (Table 3), has been found to have over 45 perfect matches with plant viral genomes (Perez-Quintero et al. 2010).
Variety-specific miRNAs
We detected miRNA160, 414, 828, 858, 3636 solely, or with remarkably more reads, in variety Chardonel (VFM411, LBC0903, SHC0903). On the other hand, miRNA393, 1520, 5021 were found only in the variety Vidal Blanc (SHV0904; Table 5). miRNA414 potentially targets eight grapevine genes while miRNA5021 targets three genes (Table 6). The prediction of the target gene’s function suggested that they are involved in multiple pathways. It remains to be determined if these miRNAs are associated specifically with a particular grape variety. There are more than 5,000 existing grape varieties in the world. The diversity of grape varieties has been studied extensively at genetic and genomic levels (Myles et al. 2011), the epigenetic diversity of grapevines remains unexplored. The revelation of variety-specific miRNAs in grapevine implies that profiles of miRNAs can be employed as a biomarker that is tagged to each specific grape variety and likely is linked with the unique characters of the grape variety.
In most studies, levels of miRNA abundance are verified by Northern blot assay or qPCR. On the other hand, the mere presence of miRNA in these samples as detected by the high throughput Solexa sequencing implies the biological significance of these miRNA in grapevines’ response to virus infection and in the development of phenotypic traits in vineyards. Among the new miRNAs, few are expressed at substantially high levels.
Conclusion
Discovery and analysis of virus infection-associated miRNAs and variety-specific miRNAs brings new perspectives on the understanding of grapevine–virus interaction at molecular levels. Infection of grapevines by viruses of different families allows us to discover novel miRNAs in grapevines and investigate their roles in grapevine’s defense response to pathogens.
References
Amin I, Patil B, Briddon R, Mansoor S, Fauquet C (2011) Comparison of phenotypes produced in response to transient expression of genes encoded by four distinct begomoviruses in Nicotiana benthamiana and their correlation with the levels of developmental miRNAs. Virol J 8(1):238
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233
Bazzini AA, Hopp HE, Beachy RN, Asurmendi S (2007) Infection and coaccumulation of tobacco mosaic virus proteins alter microRNA levels, correlating with symptom and plant development. Proc Natl Acad Sci USA 104(29):12157–12162
Bazzini A, Almasia N, Manacorda C, Mongelli V, Conti G, Maroniche G, Rodriguez M, Distefano A, Hopp HE, del Vas M et al (2009) Virus infection elevates transcriptional activity of miR164a promoter in plants. BMC Plant Biol 9(1):152
Buhtz A, Pieritz J, Springer F, Kehr J (2010) Phloem small RNAs, nutrient stress responses, and systemic mobility. BMC Plant Biol 10(1):64
Carra A, Mica E, Gambino G, Pindo M, Moser C, Pè ME, Schubert A (2009) Cloning and characterization of small non-coding RNAs from grape. Plant J 59:750–763
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136(4):642–655
Dunoyer P, Schott G, Himber C, Meyer D, Takeda A, Carrington JC, Voinnet O (2010) Small RNA duplexes function as mobile silencing signals between plant cells. Science 328(5980):912–916
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL et al (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2(2):e219
Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, Smith LM, Dasenko M, Givan SA, Weigel D et al (2010) MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22(4):1074–1089
Feng J, Lai L, Lin R, Jin C, Chen J (2012) Differential effects of Cucumber mosaic virus satellite RNAs in the perturbation of microRNA-regulated gene expression in tomato. Mol Biol Rep 39:775–784
Fung RWM, Gonzalo M, Fekete C, Kovacs LG, He Y, Marsh E, McIntyre LM, Schachtman DP, Qiu WP (2008) Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol 146:236–249
Gournas C, Papageorgiou I, Diallinas G (2008) The nucleobase–ascorbatetransporter (NAT) family: genomics, evolution, structure–function relationships and physiological role. Mol Biosyst 4:404–416
Hu Q, Hollunder J, Niehl A, Karner CJ, Gereige D, Windels D, Arnold A, Kuiper M, Vazquez F, Pooggin M et al (2011) Specific impact of Tobamovirus infection on the Arabidopsis small RNA profile. PLoS One 6(5):e19549
Jaillon O, Aury J, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C et al (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Katiyar-Agarwal S, Jin H (2010) Role of small RNAs in host–microbe interactions. Annu Rev Phytopathol 48:225–246
Kawashima CG, Matthewman CA, Huang S, Lee B-R, Yoshimoto N, Koprivova A, Rubio-Somoza I, Todesco M, Rathjen T, Saito K et al (2011) Interplay of SLIM1 and miR395 in the regulation of sulfate assimilation in Arabidopsis. Plant J 66(5):863–876
Liang G, Yang F, Yu D (2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J 62(6):1046–1057
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ et al (2008) Criteria for annotation of plant microRNAs. Plant Cell 20(12):3186–3190
Mica E, Piccolo V, Delledonne M, Ferrarini A, Pezzotti M, Casati C, Del Fabbro C, Valle G, Policriti A, Morgante M et al (2010) Correction: high throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera. BMC Genomics 11(1):109
Molnar A, Melnyk CW, Bassett A, Hardcastle TJ, Dunn R, Baulcombe DC (2010) Small silencing RNAs in plants are mobile and direct epigenetic modification in recipient cells. Science 328(5980):872–875
Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V (2008) A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 24:2252–2253
Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK, Prins B, Reynolds A, Chia J-M, Ware D et al (2011) Genetic structure and domestication history of the grape. Proc Natl Acad Sci USA 108(9):3530–3535
Naqvi A, Haq Q, Mukherjee S (2010) MicroRNA profiling of tomato leaf curl new delhi virus (tolcndv) infected tomato leaves indicates that deregulation of mir159/319 and mir172 might be linked with leaf curl disease. Virol J 7(1):281
Olsen AN, Ernst HA, Leggio LL, Skriver K (2005) NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci 10(2):79–87
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J (2010) Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J 62(6):960–976
Perez-Quintero A, Neme R, Zapata A, Lopez C (2010) Plant microRNAs and their role in defense against viruses: a bioinformatics approach. BMC Plant Biol 10(1):138
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Sunkar R, Zhu J (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16(8):2001–2019
Sunkar R, Chinnusamy V, Zhu J, Zhu J-K (2007) Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci 12(7):301–309
Tagami Y, Inaba N, Kutsuna N, Kurihara Y, Watanabe Y (2007) Specific enrichment of miRNAs in Arabidopsis thaliana infected with tobacco mosaic virus. DNA Res 14(5):227–233
Trindade I, Capitão C, Dalmay T, Fevereiro M, Santos D (2010) miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231(3):705–716
Varallyay E, Valoczi A, Agyi A, Burgyan J, Havelda Z (2010) Plant virus-mediated induction of miR168 is associated with repression of ARGONAUTE1 accumulation. EMBO J 29(20):3507–3519
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S, Reid J et al (2007) A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2(12):e1326
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136(4):669–687
Wang C, Wang X, Kibet NK, Song C, Zhang C, Li X, Han J, Fang J (2011a) Deep sequencing of grapevine flower and berry short RNA library for discovery of novel microRNAs and validation of precise sequences of grapevine microRNAs deposited in miRBase. Physiol Plant 143:64–81
Wang L, Gu X, Xu D, Wang W, Wang H, Zeng M, Chang Z, Huang H, Cui X (2011b) miR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J Exp Bot 62(2):761–773
Yoo SY, Kim Y, Kim SY, Lee JS, Ahn JH (2007) Control of flowering time and cold response by a NAC-domain protein in Arabidopsis. PLoS One 2(7):e642
Zhang X, Yuan Y-R, Pei Y, Lin S-S, Tuschl T, Patel DJ, Chua N-H (2006) Cucumber mosaic virus-encoded 2b suppressor inhibits Arabidopsis Argonaute1 cleavage activity to counter plant defense. Genes Dev 20(23):3255–3268
Zhang Y, Singh K, Kaur R, Qiu W (2011a) Association of a novel DNA virus with the grapevine vein-clearing and vine decline syndrome. Phytopathology 9:1081–1090
Zhang X, Zou Z, Gong P, Zhang J, Ziaf K, Li H, Xiao F, Ye Z (2011b) Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol Lett 33(2):403–409
Zhao M, Ding H, Zhu J, Zhang F, Li WX (2011) Involvement of miRNA169 in the nitrogen-starvation responses in Arabidopsis. New Phytol 190:906–915
Zhu C, Ding Y, Liu H (2011) miRNA398 and plant stress responses. Physiol Plant. doi:10.1111/j.1399-3054.2011.01477.x
Acknowledgments
This project was supported by USDA-CSREES (2009-38901-19962) grant to WQ. We thank Chin-Feng Hwang for critical reviewing of the manuscript. We are indebted to Brian Dalley in the Microarray and Genomic Analysis Core Facility at the University of Utah, Salt Lake City, UT, USA, for technical assistance with construction of small RNA libraries and deep sequencing.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Table 1
List of new miRNAs and their predicted target genes (DOCX 25 kb)
Supplemental Table 2
Read count of novel miRNA in each grapevine and their detailed annotations and predicted target genes (XLSX 30 kb)
Supplemental Table 3
Predicted target genes of virus-induced miRNAs in grapevine (DOCX 24 kb)
Rights and permissions
About this article
Cite this article
Singh, K., Talla, A. & Qiu, W. Small RNA profiling of virus-infected grapevines: evidences for virus infection-associated and variety-specific miRNAs. Funct Integr Genomics 12, 659–669 (2012). https://doi.org/10.1007/s10142-012-0292-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-012-0292-1