
Abstract
We acquired more than 4 million useful sequences using a high-throughput method from a library for miRNA identification, which is constructed from a mixture of 14 RNA samples from different developmental stages. We mapped 247,410 reads to known silkworm miRNAs in miRBase (13.0), 701,913 reads to other RNA molecules based on sequence homology, and 3,219,395 reads to the silkworm genome. Our analysis identified 54 silkworm known miRNAs. A striking strand bias between miRNAs and their corresponding miRNA*s was found, and was speculated to reflect that transcripts from the passenger strand of pre-miRNAs may have important biological roles. Using an elaborate screening protocol, we predicted 287 candidate novel miRNAs (represent 116,494 short reads), and 59 of them have both miRNA and miRNA* sequences. Most of the previously identified silkworm miRNAs are cross-species conserved with a high abundance, while those predicted candidates tend to be species-specific miRNAs. Our discovery of SNPs among miRNAs implied within-species functional diversity. Target prediction uncovers that considerable silkworm miRNAs may aim at modulating more than one hormone signaling pathway components and/or hormone biosynthesis-related proteins implying their important roles in silkworm development.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MicroRNAs are ∼22-nt endogenously initiated non-coding RNA (ncRNA) species that regulate the expression of target mRNAs in post-transcriptional level (Bartel 2004; Kim 2005). Increasing evidence reveals that miRNAs play significant roles in various physiological and pathological processes including development, differentiation, apoptosis, anti-viral defense, and tumorigenesis (Davison et al. 2006; Schickel et al. 2008; Wang et al. 2007). In general, miRNAs are produced from a primary stem-loop structure in nucleus and become functional after step-by-step processing in cytoplasm. The functional miRNAs have to integrate into multiprotein effector complexes, named as RNA-induced silence complex (RISC), to effectively catalyze sequence-dependent cleavage or specific repression of target mRNAs by binding to their 3′UTR regions (Denli et al. 2004; Gregory et al. 2004). Together with other non-coding RNA families, miRNA may be of great importance in articulating gene expression in details (Oulas et al. 2009).
As one of the most important economic insects, silkworm genome has been reported in 2004 (Xia et al. 2004), which provides opportunities for a thorough survey for miRNAs and their functionality. To date, only a few silkworm miRNAs are deposited in miRBase (version 13.0; http://microrna.sanger.ac.uk), and the collection is far from complete as compared with other model insects such as Drosophila. Traditional methods hold their own merits and limitations; for instance, direct cloning is superior for testing highly abundant miRNAs but not those low abundant ones and it is very expensive (Yin et al. 2008). While microarray is a powerful tool for detecting conserved miRNAs in different species and has made important contributions for large dataset surveys and specialized clinical applications, it is sensitive to background noise (Lehmussola et al. 2006). Given the complexity of the regulatory roles played by miRNAs in gene expression, their thorough discovery is of high importance, especially for the low abundance and transiently expressed miRNAs whose expression are often stringently regulated and tissue/developmental stage-specific. The SOLiD sequencing system with a sequence-by-ligation approach provides a powerful small RNA profiling method with high-throughput and accuracy.
Over 20 million raw sequence reads were obtained in the present study and analysis of the genome-matched reads yielded 287 novel candidate silkworm miRNAs, including both miRNA and miRNA* sequences (21%). Several candidates were experimentally validated by stem-loop RT PCR. We also systematically analyzed the potential SNPs and targets of silkworm miRNAs. The identification of silkworm miRNAs and the analysis of their potential targets have demonstrated that miRNAs may play an important role in the development of Bombyx mori, especially in regulating the ecdysone and juvenile hormone signaling pathways.
Materials and methods
Total RNA extraction
We collected tissue samples from silkworms (Dazao) at 14 different developmental stages demonstrated previously (Yu et al. 2008). Total RNA is extracted by using TriPure Isolation Reagent (Roche) according to manufacturer’s protocol.
Library construction and bioinformatics analysis
We isolated small RNA fractions (less than 40-nt) using the FlashPAGETM Fractionator system (Ambion, Austin, TX) and cloned them by SOLiDTM Small RNA Expression Kit (Applied Biosystems, Foster City, CA; Goff et al. 2009) according to the manufacturer's instructions. The sequencing was carried out by SOLiDTM System 2.0.
We used RNA2MAP (RNA_pipeline_0.4.0; http://solidsoftwaretools.com/gf/project/rna2map/) for primary data analysis. We first filtered reads against a dataset containing silkworm Unigene, tRNA, rRNA, Rfam sequences (excluding known miRNAs), and a custom-curated insect ncRNA database. We then attempted to align the remaining raw reads to all 55 known pre-miRNA sequences in miRBase Version 13.0(http://microrna.sanger.ac.uk/; Griffiths-Jones et al. 2008) and the sequences of the miRNA–miRNA* pairs with a 4-nt extension toward both directions.
To predict novel miRNAs, we aligned the yet unmapped reads against the genome sequences (Version 2.0; http://silkworm.genomics.org.cn/; Xia et al. 2008) by setting a 28-nt scanning window and extracted the matches with 100-nt flanking sequences for hairpin structure prediction. We used RNAfold (Denman 1993) for the prediction of candidate novel miRNAs as depicted before (Ambros et al. 2003; Yu et al. 2009).
The most abundant reads from predicted pre-miRNAs were considered as candidate mature miRNAs. After a relatively stringent screening, those with five or more copies in frequency and 25-nt or less in length were considered as candidates of novel miRNAs. The predictions were mapped to the genome for cluster analysis and cross-species comparison against other insect miRNAs and genome sequences involving 12 Drosophila species (http://flybase.org), Anopheles gambiae (http://agambiae.vectorbase.org/index.php), Apis mellifera (http://www.hgsc.bcm.tmc.edu/project-species-i-Apis%20mellifera.hgsc?pageLocation=Apismellifera), and Tribolium castaneum (http://www.hgsc.bcm.tmc.edu/projects/tribolium).
Stem-loop RT PCR
The stem-loop RT PCR experiments are carried out as described before (Chen et al. 2005). All of the designed stem-loop RT primers and gene-specific primers are listed in Supplemental file 1.
Target prediction
We used miRanda (Enright et al. 2003; John et al. 2004) version 3.1 to predict potential targets on 3’-UTR sequences retrieved from silkworm (Dazao) unigenes (ftp://ftp.ncbi.nih.gov/repository/UniGene). The thresholds for candidate target sites were S ≥ 90 and ∆G < −17 kcal/mol (Enright et al. 2003). Those silkworm miRNAs are also scanned against 3′-UTR datasets of Drosophila melanogaster and A. mellifera searching for potential targets. A target of B. mori is considered to be conserved if its orthologous in D. melanogaster or A. mellifera is also predicted to be candidate targets.
Results
Library construction, sequencing and preliminary miRNA identification
We obtained over 20 million raw reads by SOLiD sequencing from a RNA library that is constructed by using small RNA enriched samples from 14 different developmental stages. Our data processing pipeline is shown in Supplemental file 2. Since the decoding of SOLiD sequencing reads is a reference-based methodology at the present stage, we currently rely on database registries for contaminates elimination, known silkworm miRNA identification and genome matching. First, reads corresponding to rRNA, tRNA, sno/snRNA, mRNA, and other non-coding RNAs are removed; 701,913 reads that match this filter with up to one mismatch. Second, the remaining reads are used as an input file for known silkworm miRNA identification, which involves 247,410 hits with up to two mismatches. Third, the reads that have not mapped in the first filtering step or against known silkworm miRNAs are mapped to the silkworm genome in an attempt to identify novel miRNAs. In this final step, 3,219,395 reads were successfully mapped. And these reads matched to the silkworm genome are collected together with 100-nt flanking sequences for candidate miRNA prediction. Finally, in order to guarantee accuracy, we did not attempt to map more reads that do not hit any of the references using looser mapping stringency as they may contain sequencing errors, contaminants or un-sequenced/assembled genomic regions. Taken together, we successfully annotated over 4 million reads.
Known silkworm miRNAs and their abundances
In our library, we detected all of the 55 previously annotated B. mori miRNAs (miRbase Release 13.0) with the exception of bmo-miR-190 (ESM Table 1). This single failure is probably the consequence of several chance events including incompleteness of the sampling (referring to the entire silkworm life span, for instance), the delicate RNA processing procedure, and uneven sequencing depth (Fahlgren et al. 2009). Of the 54 miRNAs identified, 50 are found with both miRNA and miRNA*, and four are accounted for those with either miRNA or miRNA*. Of the 247,410 reads that map to known silkworm miRNAs, 10.4% match the hairpin structure in out of the miRNA and miRNA* regions at a high frequency consistent with the previously reported study (Ruby et al. 2006). It is believed that high-throughput sequencing not only is capable of identifying miRNAs but also provides information on gene expression ('t Hoen et al. 2008) albeit with variable results (Kato et al. 2009). Some miRNAs, such as bmo-bantam, bmo-miR-1 and bmo-miR-263a, are found at a very high frequency (∼10,000×) which alludes to their functional roles in regulating silkworm development (ESM Table 1). However, there are also many miRNAs expressed at relatively low levels, probably representing stringent transient and spatiotemporal expression. Since we constructed a library with pooled RNA samples, it is not possible to come to a conclusion of which stage these miRNAs expressed and accumulated. We also discovered a notable strand bias between miRNAs and their corresponding miRNA*s in known silkworm miRNAs (ESM Table 1) as reported previously in the case of D. melanogaster (Okamura et al. 2008). There may be functional implications in such a bias. In two such examples, bmo-miR-31 and bmo-miR-993a, whose miRNA*s are more abundant than their miRNAs, it is likely that the complementary miRNA*s are more active than their miRNAs.
Novel miRNAs and their cross-species conservation
In a search for novel candidates, we evaluated reads with perfect match to the silkworm genome that fell within miRNA-like hairpin structures, and a total of 2,036 qualified hairpins represented by 1,306,704 short reads were identified (see details in the “Materials and Methods” section). Comparison with a recent silkworm miRNA identification study based on the sequencing-by-synthesis method reveals an overlap of 16 miRNAs between the two datasets (Zhang et al. 2009). Besides, these candidate novel miRNAs also included six overlaps with our former discoveries published recently (Supplemental file 3) (Yu et al. 2009). We obtained 287 novel miRNA candidates mapped with 116,494 short reads in their stem regions(provisional named with prefix bmo-miR-p), in which about 20% of candidates contain both miRNA and miRNA* sequences (Table 1) and the remaining reads were presented either in miRNAs or miRNA*s (Supplemental files 4 and 5). We believe that the detection of miRNA*s is a strong clue, albeit not absolute, for the formation of precursor hairpin structures and adds weight to the authenticity of the predicted candidates (Fahlgren et al. 2007; Sunkar et al. 2008). These novel candidates displayed a concentrated length distribution between 21 and 25 nt with a peak at ∼23 nt as well as a robust U bias at 5′ ends, which is a characteristic of miRNAs produced from dsRNA precursors by Dicer. To confirm the expression of the identified miRNAs, we performed stem-loop RT PCR and validated 36, out of 43 attempted, of the candidate novel B. mori miRNA sequences (Fig. 1). The remaining seven candidates were excluded due to non-specific amplification.

Stem-loop RT PCR. Experimental validation of a subset of the predicted miRNAs
To determine whether the novel candidates are conserved among other insect species, we searched their sequences against other insect miRNAs and genome sequences, including T. castaneum, A. gambiae, A. mellifera, and twelve Drosophila species. As a result, 81 silkworm candidates are identified as cross-species conserved with high confidence; there are eight homologs that are already reported in other insects (bmo-miR-316, bmo-miR-1000, bmo-miR-998, bmo-miR-996, bmo-miR-11, bmo-miR-308, bmo-miR-306, and bmo-miR-1175; directly named according to their homologs). The other 73 candidates are mapped to at least one or more insect genomes and foldable into eligible hairpin structures (Supplemental file 6). The remaining candidates do not have cross-species homologs, which probably represent silkworm-specific miRNAs.
SNPs in the silkworm miRNAs
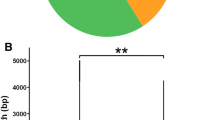
We also found single-nucleotide polymorphism phenomena in our miRNA library. We identified SNP sites in both “seed” region and outside of the “seed” region on the mature miRNAs (Fig. 2a). Most of the SNP sites are located in the outside of the “seed” region suggest that the “seed” region of miRNA is more conservative than the other region of miRNA. We failed to recognize an obvious SNP bias in the known and the novel candidate miRNAs because of the arbitrary distribution of the observed SNPs. Although some of the SNPs in mature miRNAs may be false positive resulting from assembling or sequencing errors, we believe that the highly abundant ones may come from individual differences at DNA level, which need to be further validated and distinguished from post-transcriptional modification events, such as miRNA editing (Ohman 2007).

Possible SNPs and clusters in silkworm miRNAs. a The most representative single-nucleotide polymorphisms in the known miRNAs and the new candidate miRNAs in silkworm are illustrated. The mature miRNAs, hairpin locations, and putative SNPs are in green, yellow, and red, respectively. The folding energy (dG), mature miRNA length, and frequencies are also shown. b Candidate novel miRNAs located in cluster structures are shown in appropriate direction and color-coded (not drawn to scale)
Cluster analysis
One of the typical characteristics of miRNA is that closely juxtaposed miRNA genes are inclined to transcribe from a common promoter to generate polycistronic primary structures (Baskerville and Bartel 2005; Lee et al. 2004). In our study, 31 of the novel miRNA candidates are also grouped into 13 clusters; most of them are composed of two miRNAs (Supplemental file 7). The top cluster is made up of six miRNA molecules, and each has a high detection frequency (Fig. 2b). Surprisingly, some of the previously annotated miRNAs located in cluster structures that are proposed to co-express showed a strong inconsistency in our results. For example, the empirical expression level of bmo-miR-1 is nearly forty times higher than that of bmo-miR-133 and the two are 16 kb apart in B. mori. The reason for this disparity remains unknown.
Potential targets for silkworm miRNAs
The expression of miRNAs is generally considered to be negatively correlated with their mRNA targets. To gain an overview on the functions of the silkworm miRNAs, we performed miRanda (Enright et al. 2003) to predict potential targets. The thresholds for candidate target sites were S ≥ 90 and ∆G < −17 kcal/mol, and those unqualified targets are not considered for further analysis. Silkworm miRNAs are scanned against 3′-UTR datasets of B. mori, D. melanogaster, and A. mellifera, respectively, for candidate targets prediction. As a result, we obtained 172 conserved targets that involved in a broad range of biological functions, such as transcription factors, signal transduction, metabolism enzymes, and cancer-related genes (Supplemental file 8). Many genes involved in several levels of the ecdysone cascades and the downstream transcription factors are predicted, including EcR, Broad-Complex, pre-prothoracicotropic hormone, nuclear receptor HR3 and nuclear orphan receptors(data not shown). We also obtained several target genes that are involved in the biosynthesis and metabolism of ecdysone such as cytochrome P450, ecdysteroid-phosphate phosphatase and ecdysone-20-hydroxylase.
Discussion
The miRNAs have been generally considered as negative regulators of gene expression in cellular. Identifying the total number of miRNA genes in a species, especially those low-abundance and species-specific ones, is helpful for appreciating the breadth of miRNA functions. MiRNAs have been identified in diverse animal species, and the large-scale miRNA identification in insects with experimental approaches has mainly been performed in fly. Herein, we constructed, sequenced, and analyzed a small RNA library made from a pooled RNA samples from multi-developmental stages of the domesticated silkworm (Dazao) based on the SOLiD high-throughput sequencing system. In total, we identified 287 novel miRNA candidates, in which 59 of the candidates contain both miRNA and miRNA* sequences. Eight of the 287 novel candidates are already reported in other insects. We show that the method is effective in identifying low-abundance and species-specific small RNAs. Additionally, we experimentally verified 36 of the candidate novel miRNAs. Compared to the previously identified silkworm miRNAs, the abundance of predicted novel miRNAs are much lower as indicated by their frequencies, which may account for tissue- or species-specific expression. And, as expected, most of the candidate novel miRNAs are not cross-species conserved. However, it is also likely that several novel non-coding RNA molecules may exist, generating analogous forms to miRNAs by forming hairpin structures, such as endogenous siRNAs, which cannot be excluded due to the lack of database information. All together, deep sequencing is a rapid and effective approach to uncover rare and species-specific miRNAs and the identification of a near complete set of miRNAs for a species is of fundamental importance to establish a basis for unraveling the complex miRNA-mediated regulatory networks.
We noticed a strand bias between miRNAs and their corresponding miRNA*s in known silkworm miRNAs. In the cases of bmo-miR-31 and bmo-miR-993a, the miRNA* strands are more abundant than their miRNA strands, it is likely that the complementary miRNA*s are more active than their miRNAs and thus maintaining a high concentration. Recently, reports sprang up and suggested that transcripts from the passenger strand of pre-miRNAs can also profoundly affect many biological pathways. Kim et al. demonstrated that miR-199a* is able to target MET (mesenchymal–epithelial transition factor) proto-oncogene and its downstream effector ERK2 (extracellular signal-regulated kinase 2) and inhibited the proliferation, motility and invasive capabilities of tumor cells (Kim et al. 2008). In the case of pre-miR-146a, Jazdzewski et al. proposed that miR-146a* from the passenger strand may regulate many genetic processes in thyroid cancer (Jazdzewski et al. 2009).
Single-nucleotide polymorphisms (SNPs) are a novel class of functional variations that may affect the generation and function of a miRNA and result in a change of target mRNAs. Importantly, an increasing number of studies have supported an association between miRNA SNPs and tumorigenesis (Chin et al. 2008; Horikawa et al. 2008; Jazdzewski et al. 2009). We focused on the identification of SNPs in mature miRNA regions and obtained a set of SNP sites with high frequencies which increased their authenticity. A few of the SNP sites is located in the 5′ seed region that implies their conservation is relatively higher than the other parts of a mature miRNA. Recently, a growing body of work has illustrated that SNPs in the sequences flanking the pre-miRNAs may affect processing by Drosha/Dicer and result in the impaired/enhanced expression of mature miRNAs (Duan et al. 2007; Hu et al. 2008; Sun et al. 2009). In animals, the “seed” region is considered as the key binding site for target identification, and a single-nucleotide polymorphism in this region may lead to loss or gain of function and greatly change the mediated genes depending on whether they interfere, degenerate, and generate a binding site (Jazdzewski et al. 2009). In short, miRNA SNPs have brought in a new field of research in miRNA functional studies and should be taken into account in the future functional assays.
One of the typical characteristics of miRNA is that closely juxtaposed miRNA genes are inclined to transcribe from a common promoter to generate polycistronic primary structures. It was demonstrated that the secondary structure of a clustered pre-miRNA is more similar to its neighboring pre-miRNAs in the same cluster, when compared to other sequences outside clusters (Leung et al. 2008). Lately, reliable evidence has been published (Kim et al. 2009) to support the hypothesis that miRNA genes of the same cluster may play related biological functions. Young-Kook Kim and colleagues reported that miRNAs in two clusters (miR-106b ∼93∼25 and miR-222∼221) suppressed the expression of the Cip/Kip family members of Cdk inhibitors in gastric cancer tissues. The increasing amount of evidence has led us to speculate that the organization as well as the functional association of clustered miRNAs should be an effective way to coordinate genomic resources and has survived long-term natural selection.
Our work shows that most of the potential targets of silkworm miRNAs are not conserved between species and are highly enriched in ecdysone and juvenile hormone signaling pathway regulators as well as hormone metabolism-related proteins. Ecdysone signaling regulation participates in controlling various biological pathways and plays a critical role in the developmental transitions of insects (Cruz et al. 2007; Horike and Sonobe 1999). Generally, hormone signals bind to the ecdysone receptor (EcR) and trigger the regulation of the expression of different transcription factors, such as the zinc finger proteins of the Broad-Complex and other kinds of nuclear hormone receptors, which in turn modulate central regulators of different physiological processes. Conversely, juvenile hormones perform an important antagonistic function to the biological processes controlled by ecdysone and results in the maintaining of larval modality, preventing the metamorphosis and development of the adult imaginal disk (Enright et al. 2003).
Interestingly, considerable silkworm miRNAs are predicted to aim at more than one hormone signaling pathway genes and/or hormone biosynthesis-related proteins, such as EcR, Broad-Complex, nuclear receptor HR3 isoform A, cytochrome P450, ecdysteroid-phosphate phosphatase, and nuclear orphan receptors. Therefore, we are prompt to speculate that miRNAs may directly or indirectly fine-tune a series of ecdysone-biosynthesis-related genes in order to regulate various developmental states in the life cycle of the silkworms. These predictions as well as speculations only come true when supported by experimental evidences. The accumulating knowledge about miRNA function will contribute to our better understanding of the complex gene regulatory networks at both transcriptional and post-transcriptional levels.
It has been reported that the expression of some miRNAs, such as let-7 and miR-34, are affected by ecdysone in Drosophila during metamorphosis (Sempere et al. 2002; Sempere et al. 2003). Besides, let-7 has been detected to express temporally and spatially in cultured cell lines of silkworm that showed a clear association with ecdysone pulse and a variety of biological processes (Liu et al. 2007). However, the mechanism of how bmo-let-7 responds to ecdysone and other hormone pathways in silkworm is still unknown. In our study, let-7 was highly expressed (represent by 4,683 short reads) and was predicted to target juvenile hormone acid methyltransferase (JHAMT) and hemolymph juvenile hormone binding protein (hJHBP), both of which were associated with juvenile hormone activity (Niwa et al. 2008; Wieczorek et al. 1996). Besides, miR-34 were predicted to aim at several nuclear receptor superfamily members including EcR, HR3 isoform A and orphan receptor, which played key roles in ecdysone regulation mechanisms (Eystathioy et al. 2001; Sutherland et al. 1995). Further experiments of gain-of-function and loss-of-function are still needed and will determine how many of these predicted targets are genuinely and directly targeted by miRNAs in silkworm.
Reference
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, Matzke M, Ruvkun G, Tuschl T (2003) A uniform system for microRNA annotation. RNA 9:277–279
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Baskerville S, Bartel DP (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11:241–247
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179
Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA, Crowell RE, Patel R, Kulkarni T, Homer R, Zelterman D, Kidd KK, Zhu Y, Christiani DC, Belinsky SA, Slack FJ, Weidhaas JB (2008) A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res 68:8535–8540
Cruz J, Martin D, Belles X (2007) Redundant ecdysis regulatory functions of three nuclear receptor HR3 isoforms in the direct-developing insect Blattella germanica. Mech Dev 124:180–189
Davison TS, Johnson CD, Andruss BF (2006) Analyzing micro-RNA expression using microarrays. Methods Enzymol 411:14–34
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432:231–235
Denman RB (1993) Using RNAFOLD to predict the activity of small catalytic RNAs. Biotechniques 15:1090–1095
Duan R, Pak C, Jin P (2007) Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet 16:1124–1131
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS (2003) MicroRNA targets in Drosophila. Genome Biol 5:R1
Eystathioy T, Swevers L, Iatrou K (2001) The orphan nuclear receptor BmHR3A of Bombyx mori: hormonal control, ovarian expression and functional properties. Mech Dev 103:107–115
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC (2007) High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219
Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TW, Givan SA, Carrington JC (2009) Computational and analytical framework for small RNA profiling by high-throughput sequencing. RNA 15:992–1002
Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, Sun YE, Hart RP (2009) Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PLoS One 4:e7192
Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R (2004) The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–158
Horikawa Y, Wood CG, Yang H, Zhao H, Ye Y, Gu J, Lin J, Habuchi T, Wu X (2008) Single nucleotide polymorphisms of microRNA machinery genes modify the risk of renal cell carcinoma. Clin Cancer Res 14:7956–7962
Horike N, Sonobe H (1999) Ecdysone 20-monooxygenase in eggs of the silkworm, Bombyx mori: enzymatic properties and developmental changes. Arch Insect Biochem Physiol 41:9–17
Hu Z, Chen J, Tian T, Zhou X, Gu H, Xu L, Zeng Y, Miao R, Jin G, Ma H, Chen Y, Shen H (2008) Genetic variants of miRNA sequences and non-small cell lung cancer survival. J Clin Invest 118:2600–2608
Jazdzewski K, Liyanarachchi S, Swierniak M, Pachucki J, Ringel MD, Jarzab B, de la Chapelle A (2009) Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc Natl Acad Sci U S A 106:1502–1505
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS (2004) Human MicroRNA targets. PLoS Biol 2:e363
Kato M, de Lencastre A, Pincus Z, Slack FJ (2009) Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during C. elegans development. Genome Biol 10:R54
Kim VN (2005) MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 6:376–385
Kim S, Lee UJ, Kim MN, Lee EJ, Kim JY, Lee MY, Choung S, Kim YJ, Choi YC (2008) MicroRNA miR-199a* regulates the MET proto-oncogene and the downstream extracellular signal-regulated kinase 2 (ERK2). J Biol Chem 283:18158–18166
Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, Hur K, Yoo MW, Lee HJ, Yang HK, Kim VN (2009) Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res 37:1672–1681
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Lehmussola A, Ruusuvuori P, Yli-Harja O (2006) Evaluating the performance of microarray segmentation algorithms. Bioinformatics 22:2910–2917
Leung WS, Lin MC, Cheung DW, Yiu SM (2008) Filtering of false positive microRNA candidates by a clustering-based approach. BMC Bioinformatics 9(Suppl 12):S3
Liu S, Xia Q, Zhao P, Cheng T, Hong K, Xiang Z (2007) Characterization and expression patterns of let-7 microRNA in the silkworm (Bombyx mori). BMC Dev Biol 7:88
Niwa R, Niimi T, Honda N, Yoshiyama M, Itoyama K, Kataoka H, Shinoda T (2008) Juvenile hormone acid O-methyltransferase in Drosophila melanogaster. Insect Biochem Mol Biol 38:714–720
Ohman M (2007) A-to-I editing challenger or ally to the microRNA process. Biochimie 89:1171–1176
Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC (2008) The regulatory activity of microRNA* species has substantial influence on microRNA and 3' UTR evolution. Nat Struct Mol Biol 15:354–363
Oulas A, Boutla A, Gkirtzou K, Reczko M, Kalantidis K, Poirazi P (2009) Prediction of novel microRNA genes in cancer-associated genomic regions—a combined computational and experimental approach. Nucleic Acids Res 37:3276–87
Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP (2006) Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127:1193–1207
Schickel R, Boyerinas B, Park SM, Peter ME (2008) MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 27:5959–5974
Sempere LF, Dubrovsky EB, Dubrovskaya VA, Berger EM, Ambros V (2002) The expression of the let-7 small regulatory RNA is controlled by ecdysone during metamorphosis in Drosophila melanogaster. Dev Biol 244:170–179
Sempere LF, Sokol NS, Dubrovsky EB, Berger EM, Ambros V (2003) Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Dev Biol 259:9–18
Sun G, Yan J, Noltner K, Feng J, Li H, Sarkis DA, Sommer SS, Rossi JJ (2009) SNPs in human miRNA genes affect biogenesis and function. RNA 15:1640–51
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK (2008) Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol 8:25
Sutherland JD, Kozlova T, Tzertzinis G, Kafatos FC (1995) Drosophila hormone receptor 38: a second partner for Drosophila USP suggests an unexpected role for nuclear receptors of the nerve growth factor-induced protein B type. Proc Natl Acad Sci U S A 92:7966–7970
T Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT (2008) Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res 36:e141
Wang Y, Stricker HM, Gou D, Liu L (2007) MicroRNA: past and present. Front Biosci 12:2316–2329
Wieczorek E, Parkitna JM, Szkudlarek J, Ozyhar A, Kochman M (1996) Immunoaffinity purification of juvenile hormone-binding protein from Galleria mellonella hemolymph. Acta Biochim Pol 43:603–610
Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, Pan G, Xu J, Liu C, Lin Y, Qian J, Hou Y, Wu Z, Li G, Pan M, Li C, Shen Y, Lan X, Yuan L, Li T, Xu H, Yang G, Wan Y, Zhu Y, Yu M, Shen W, Wu D, Xiang Z, Yu J, Wang J, Li R, Shi J, Li H, Su J, Wang X, Zhang Z, Wu Q, Li J, Zhang Q, Wei N, Sun H, Dong L, Liu D, Zhao S, Zhao X, Meng Q, Lan F, Huang X, Li Y, Fang L, Li D, Sun Y, Yang Z, Huang Y, Xi Y, Qi Q, He D, Huang H, Zhang X, Wang Z, Li W, Cao Y, Yu Y, Yu H, Ye J, Chen H, Zhou Y, Liu B, Ji H, Li S, Ni P, Zhang J, Zhang Y, Zheng H, Mao B, Wang W, Ye C, Wong GK, Yang H (2004) A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306:1937–1940
Xia Q et al (2008) The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol 38:1036–1045
Yin JQ, Zhao RC, Morris KV (2008) Profiling microRNA expression with microarrays. Trends Biotechnol 26:70–76
Yu X, Zhou Q, Cai Y, Luo Q, Lin H, Hu S, Yu J (2009) A discovery of novel microRNAs in the silkworm (Bombyx mori) genome. Genomics 94:438–444
Yu X, Zhou Q, Li SC, Luo Q, Cai Y, Lin WC, Chen H, Yang Y, Hu S, Yu J (2008) The silkworm (Bombyx mori) microRNAs and their expressions in multiple developmental stages. PLoS ONE 3:e2997
Zhang Y, Zhou X, Ge X, Jiang J, Li M, Jia S, Yang X, Kan Y, Miao X, Zhao G, Li F, Huang Y (2009) Insect-specific microRNA involved in the development of the silkworm Bombyx mori. PLoS One 4:e4677
Acknowledgments
We would like to thank Professor Anying Xu of the Sericultural Research Institute, Chinese Academy of Agricultural Sciences, for providing silkworm eggs. We are especially grateful for the support of bioinformatics analysis from Jiandong Sun (Life Technologies, Inc.). This work was supported by the Knowledge Innovation Program of the Chinese Academy of Sciences (08SQN01185) awarded to Xiaomin Yu, and a grant from the Ministry of Science and Technology(2006CB910400) awarded to Jun Yu.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yimei Cai, Xiaomin Yu, and Qing Zhou contributed equally to this work.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Supplemental file S1
Designed stem-loop RT PCR primers. (XLS 35 kb) (XLS 35 kb)
Supplemental file S2
Data processing pipeline. (GIF 19 kb)
Supplemental file S3
Overlaps with published miRNA data. (DOC 45 kb) (DOC 45 kb)
Supplemental file S4
List of silkworm candidate novel miRNAs without stars. (XLS 46 kb) (XLS 46 kb)
Supplemental file S5
Candidate novel silkworm miRNA hairpin structures. (TXT 94 kb) (TXT 94 kb)
Supplemental file S6
Cross-species conserved candidate novel miRNAs in other insects. (XLS 69 kb) (XLS 69 kb)
Supplemental file S7
Genome location of the pre-miRNA sequences. (XLS 97 kb) (XLS 97 kb)
Supplemental file S8
Predicated targets with species conservation. (XLS 96 kb) (XLS 96 kb)
ESM Table 1
Details about known silkworm miRNAs identified by SOLiD sequencing. (DOC 100 kb)
Rights and permissions
About this article
Cite this article
Cai, Y., Yu, X., Zhou, Q. et al. Novel microRNAs in silkworm (Bombyx mori). Funct Integr Genomics 10, 405–415 (2010). https://doi.org/10.1007/s10142-010-0162-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-010-0162-7