
Abstract
The silk gland of the silkworm Bombyx mori is a specialized organ where silk proteins are efficiently synthesized under precise regulation that largely determines the properties of silk fibers. To understand the genes involved in the regulation of silk protein synthesis, considerable research has focused on the transcripts expressed in silk glands; however, the complete transcriptome profile of this organ has yet to be elucidated. Here, we report a full-length silk gland transcriptome obtained by PacBio single-molecule long-read sequencing technology. In total, 11,697 non-redundant transcripts were identified in mixed samples of silk glands dissected from larvae at five developmental stages. When compared with the published reference, the full-length transcripts optimized the structures of 3002 known genes, and a total of 9061 novel transcripts with an average length of 2171 bp were detected. Among these, 1403 (15.5%) novel transcripts were computationally revealed to be lncRNAs, 8135 (89.8%) novel transcripts were annotated to different protein and nucleotide databases, and 5655 (62.4%) novel transcripts were predicted to have complete ORFs. Furthermore, we found 1867 alternative splicing events, 2529 alternative polyadenylation events, 784 fusion events and 6596 SSRs. This study provides a comprehensive set of reference transcripts and greatly revises and expands the available silkworm transcript data. In addition, these data will be very useful for studying the regulatory mechanisms of silk protein synthesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The silk produced by the domesticated silkworm Bombyx mori is an excellent natural protein that has been extensively used not only in the textile industry but also in the fields of tissue engineering, biomaterials, and cosmetics, among others (Song et al. 2016; Li et al. 2015; Omenetto and Kaplan 2010). The silk gland of B. mori, which is morphologically divided into the anterior silk gland (ASG), middle silk gland (MSG) and posterior silk gland (PSG), is a highly specialized organ that controls the synthesis of two major components of silk protein: the fibroin protein, which is synthesized in the PSG, and the sericin protein, which is synthesized in the MSG. In the last larval instar (the fifth larval instar, 5L), especially after the third day of 5L, the silk gland grows rapidly and synthesizes a remarkable quantity of silk proteins; fibroin is continuously transported out of the PSG, accumulated and coated with sericin in the MSG, and finally secreted though the ASG to form the cocoon (Tomita 2011; Takasu et al. 2010). The mechanism by which silk proteins are efficiently synthesized and precisely regulated is an important but challenging research topic, as silk protein synthesis is a complex genetic trait in B. mori.
Previous studies have shown that silk protein synthesis is regulated mainly at the transcriptional level, and numerous genes are involved in this process (Couble et al. 1987; Obara and Suzuki 1988). To understand which genes are associated with silk protein synthesis, efforts have been made to reveal the silk gland transcriptome. For example, Zhong et al. (2005) identified 2861 consistent expressed sequence tags (EST) from the PSG of fifth-instar larvae and found that most of them functioned in fibroin synthesis and secretion. Xia et al. (2008) designed a whole-genome oligonucleotide microarray and identified 4432 and 4269 active genes from the ASG/MSG and PSG of day-3 fifth-instar larvae, respectively. Royer et al. (2011) compared the transcriptomes of the MSG and PSG using the serial analysis of gene expression (SAGE) method. Fang et al. (2015) compared the silk gland transcriptomes of domestic and wild silkworms. Wang et al. (2016a, b) identified 630 novel transcripts from the PSG of the ZB strain, which has low silk production, and most of them were upregulated compared with those in the control strain. Cui et al. (2018) analyzed the transcriptomes of the MSG and PSG of day-4 fifth-instar larvae of the fibroin heavy chain (fibH) knockout mutant and wild type and identified 1456 differentially expressed genes (DEGs) between the PSG and MSG and 1388 DEGs between the mutant and the wild type. Shiet al. (2019) conducted a comparative transcriptome analysis of seven segments of a single silk gland and identified 3121 DEGs among these segments. Hu et al. (2019) compared the PSG transcriptomes of the Nd mutant and wild type and identified 2178 DEGs between them. These studies and their associated sequencing data have provided a general overview of silk protein synthesis. However, it remains challenging to fully elucidate the roles of all the genes involved in silk protein synthesis due to the complexity of gene expression and regulation. Another challenge is to reliably assemble full-length transcripts from the short reads generated using different second-generation sequencing (SGS) methods, which could result in the loss of some important information and prevent accurate evaluation of long transcripts, repetitive sequences, transposable elements, etc. (Michael and VanBuren 2015). Therefore, clarifying the complexity of the silk gland transcriptome requires more reliable high-throughput tools for transcriptome analysis.
Recently, a third-generation sequencing (TGS) technology called single-molecule real-time sequencing (SMRT) has been developed and used for whole-transcriptome profiling in humans, animals, and plants (Sharon et al. 2013; Yi et al. 2018; Wen et al. 2018; Kuo et al. 2017; Chen et al. 2018; Cheng et al. 2017; Wang et al. 2016a, b; Shen et al. 2014). SMRT overcomes the drawbacks of previous technologies by generating sequence information with long reads, low systematic bias, and high consensus read accuracy and is highly effective for discovering novel genes, determining allele-specific expression and unraveling transcript diversity, among other applications (Rhoads and Au 2015; Korlach et al. 2010). Here, for the first time, we used Pacific Biosciences (PacBio) SMRT sequencing to perform whole-transcriptome profiling of the silk gland of B. mori. The results will not only promote our understanding of the transcriptional regulation of silk protein synthesis but also provide a comprehensive set of reference transcripts that can greatly revise and expand the currently available silkworm transcripts.
Materials and methods
Animals and sample preparation
The domesticated silkworm strain Nistari was maintained in our laboratory and was used to collect sequencing samples. The hatched larvae were reared on fresh mulberry leaves at 25–28 °C. The silk glands were dissected from day-3 fourth-instar larvae (4L3D), day-1 fifth-instar larvae (5L1D), day-3 fifth-instar larvae (5L3D), day-5 fifth-instar larvae (5L5D), and day-6 fifth-instar larvae (5L6D), mixed equally and divided into three equal parts. Subsequently, these samples were subjected to RNA extraction using TRIzol reagent (Invitrogen, CA, USA). Total RNA was then treated with an aliquot of DNase (Takara, Dalian, China) to eliminate DNA. Both Nanodrop and Agilent 2100 instruments were used to assess RNA quality.
Library construction and sequencing
The total RNA was reverse transcribed into cDNA using the SMARTer™ PCR cDNA Synthesis Kit (Clontech Laboratories, Inc., USA) according to the official protocol. Full-length (FL) cDNAs were size selected with the BluePippin DNA Size Selection System protocol (Labtech International Ltd., England) to establish three libraries with different sizes (1–2, 2–3 and 3–6 kb in length). Using the SMRTbell Template Prep Kit, the cDNA products were then used to synthesize SMRTbell Template libraries, which were subjected to another PCR. After library quality control assessment, a total of five SMRT cells were sequenced on the PacBio RS II platform (Biomarker Technologies Corporation, Beijing, China). The raw data were uploaded to the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/) with accessions SRR10716271 and SRR10716270 (1–2 kb data), SRR10716270 and SRR10716268 (2–3 kb data), and SRR10716267 (3–6 kb data).
Analysis of full-length (FL) transcripts
The raw polymerase reads were extracted to generate reads of insert (ROIs) with full passes ≥ 0 and a predicted consensus accuracy > 0.75 that were considered FL or non-FL transcript sequences by screening for the existence of 5′/3′ cDNA primers and poly-A tails. Then, iterative clustering for error correction (ICE) was used to obtain consensus isoforms by clustering FL sequences. Finally, the Quiver algorithm was applied to cluster the non-FL sequences and polish the consistent sequences to produce high-quality (postcorrection accuracy above 99%) and low-quality transcripts. To further improve the accuracy, we used Illumina RNA-seq data to polish the low-quality isoforms and generate corrected consensus sequences. By mapping the corrected consensus sequences to the reference genome with GMAP (Wu and Watanabe 2005), reads with coverage less than 85% and identity less than 90% were removed. Reads altered only at the 5′-start site within the first exon were regarded as redundant reads and were eliminated to yield the non-redundant transcripts. The entire processing pipeline is outlined in Fig.S1.
Gene model construction
Alternative splicing (AS) analysis
The sequences of the non-redundant transcripts were validated against known B. mori reference transcript annotations with the python library MatchAnnot. Five AS types, intron retention (IR), exon skipping (ES), alternative 5′ donor sites (AD), alternative 3′ acceptor sites (AA) and mutually exclusive exons (MEE), were identified by AStalavista ( Foissac and Sammeth 2007) with the default parameters. To validate the AS events, we downloaded the Illumina RNA-Seq data from B. mori silk glands, including ASG (accession SRR4425254), MSG (accession SRR4425259), and PSG (accession SRR4425256) data, reported by Chang et al. (2015) from the SRA. The reads were then aligned against the reference gene set from KAIKObase (https://sgp.dna.affrc.go.jp/KAIKObase/) (Suetsugu et al. 2013). Miso was used to visualize the splicing events with a Sashimi plot (Katz et al. 2010, 2015).
Alternative polyadenylation (APA) analysis
Based on the full-length non-chimeric (FLNC) reads, we used the TAPIS pipeline (Abdel-Ghany et al. 2016) to identify APA with the default parameters.
Gene structure optimization
Based on the AS results, areas outside the reference gene boundaries were augmented by mapped reads that extended the untranslated regions (UTR) of the genes upstream and downstream to modify the gene boundaries with an inhouse script. Combining both the AS results and reference gene sets, we built an updated, comprehensive gene model (MergedSet). The completeness of the MergedSet was assessed by Benchmarking Universal Single-Copy Orthologs (BUSCO) (Simão et al. 2015).
Simple sequence repeat (SSR) analysis
MISA (MIcroSAtellite identification tool; Beier et al. 2017) is a program that is widely used for the identification of simple repetitive sequences. Transcripts with lengths greater than 500 bp were subjected to SSR analysis using MISA software. Seven types of SSR, namely, mononucleotide (single base), dinucleotide (two bases), trinucleotide (three bases), tetranucleotide (four bases), pentanucleotide (five bases), hexanucleotide (six bases), and compound (mixed microsatellites, two SSRs with a distance of less than 100 bp), can be detected by analyzing transcript sequences.
LncRNA prediction and target gene prediction
Four computational approaches, including CPC, CNCI, CPAT, and Pfam, were combined to sort non-protein-coding RNA candidates from putative protein-coding RNAs in the transcripts. The putative protein-coding RNAs were identified with a minimum length and exon number threshold. Transcripts with lengths greater than 200 nt and more than two exons were selected as lncRNA candidates and further screened using CPC/CNCI/CPAT/Pfam to distinguish protein-coding genes from noncoding sequences. Based on the complementary base pairing relationships between lncRNAs and mRNAs, we used LncTar software to predict the target genes of the lncRNAs.
Fusion transcript analysis
Transcripts that mapped to more than 1 locus with coverage ≥ 5% at each locus as well as total coverage ≥ 95% were extracted from the corrected consensus sequences. Subsequently, sequences with distances of over 10 kb between mapped loci or mapping to different scaffolds were selected to obtain the set of fusion transcripts with an inhouse package.
Annotation of novel transcripts
The novel non-redundant transcripts were selected, and functional annotation was conducted by using the BLAST (version 2.2.26) toolkit (Altschul et al. 1997) against a series of nucleotide and protein databases: NR (NCBI non-redundant protein sequences), Swiss-Prot (a manually annotated and reviewed protein sequence database), COG (Clusters of Orthologous Groups of proteins), Pfam (a database of conserved protein families or domains), and KEGG (Kyoto Encyclopedia of Genes and Genomes). Blast2GO was used to annotate the transcripts with Gene Ontology (GO) terms based on the NR annotations. TransDecoder software was used to predict the sequence of each coding region and its corresponding amino acid sequence.
Transcription factor (TF) analysis
We used getorf to find the ORF of each isoform and then aligned the ORF to the animal TF database (AnimalTFDB) by DIAMOND (Simão et al. 2015). Isoforms with the same TF domain were used to generate a phylogenetic tree with Muscle and MEGA (Michael and VanBuren 2015).
Results
Transcriptome sequencing of B. mori silk glands
To acquire a comprehensive full-length transcriptome of B. mori silk glands, three libraries with cDNA insert sizes of 1–2 kb, 2–3 kb, and 3–6 kb were generated based on RNAs from apooled mixture of silk glands from five larval stages. Using SMRT sequencing technology, a total of 751,460 polymerase reads were generated on five SMRT cells, and 4,627,296 subreads were obtained after removing subreads with lengths less than 50 bp or accuracies less than 0.75 (Table S1). In total, 321,648 ROIs were obtained, as shown in Tables 1 and S2, and 164,243 ROIs were identified as FLNC reads with a mean length of 1935 bp. Furthermore, 21,137 high-quality and 7670 low-quality consensus isoforms were obtained based on the clustering algorithm of ICE. After correction using ~ 1.67 Gb paired-end reads produced by Illumina sequencing, a total of 28,807 corrected isoforms with a mean length of 2263 bp were produced and could be mapped to the reference genome using GMAP. Eventually, 11,697 non-redundant transcripts were acquired (Tables 2 and S3).
Construction of comprehensive gene models
We compared 11,697 PacBio assembly isoforms (PBset, Table S3) against the reference gene annotation from KAIKObase (KAIKObaset), with 81.49% of those isoforms aligned to 5225 annotated genes. We identified 2636 isoforms concordant with KAIKObaset, 6896 novel isoforms from known genes that could optimize the 5′/3′ UTRs for 3002 known genes, and 2165 novel isoforms of novel genes (Fig. 1a). Based on coding sequence (CDS) predictions of the novel isoforms, we identified 5655 novel complete open reading frames (ORFs). Overall, these novel transcripts (mean: 2171 bp) were significantly longer than the KAIKObaset sequences (mean: 1601 bp), demonstrating the benefits of the longer read lengths of PacBioSMRT technology (Fig. 1b).

Component of assembly isoforms. a Classification of assembly isoforms mapped to the reference genome. b Length distribution of PacBio transcripts derived from known and novel genic loci
To evaluate the completeness of the transcriptome assembly, we conducted BUSCO analyses with the Insecta gene set (insecta_odb9, which contains 1658 single-copy genes that are highly conserved in insects) to assess the gene set from SilkDB v2 (Duan et al. 2010), KAIKObaset and the PBset. The PBset showed the fewest fragmented BUSCOs. Then, the PBset and KAIKObaset were merged to generate a comprehensive gene set (MergedSet), whose completeness increased remarkably to 94.8%, furthermore, lowering the number of fragmented and missing BUSCOs (Table 2). Given these results, we can conclude that this experiment established a more complete genome annotation.
Detection of AS and APA events
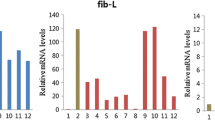
In total, 1867 AS events were detected from the PacBio de novo transcriptome of the silk gland (Table S4), including 74 MEE, 682 IR events, 551 ES events, 217 AD and 343 AA. Significantly, six major silk protein coding genes, including the fibroin protein genes fibH, fibL and P25 and the sericin protein genes Ser1, Ser2 and Ser3, were found to have seven (fibH), one (fibL), three (P25), 127 (Ser1), 11 (Ser2) and zero (Ser3) AS events (Fig. 2). This finding was quite different from that of a previous report, which indicated that only Ser1 had a variety of alternative isoforms (Garel et al. 1997); this contrast suggests the complex regulation of genes coding for silk proteins and deserves further study. AS events were also found in several crucial transcription factors that are responsible for the regulation of silk protein synthesis, including Dimm, Sage, SGF1, and SGF2/Awh (Fig. S2). In addition, we found that several genes in the 20E signaling pathway, which are crucial for regulating silk protein synthesis, harbored complex AS events (Table S5). These results suggest that AS of the above genes may contribute to the coordinated and complex regulation of silk protein synthesis, and this prospect deserves to be studied further.

AS events in major silk protein coding genes. a AS events in three fibroin protein genes. b AS events in three sericin protein genes. Sashimi plot of ES and truncation events in the ASG, middle MSG (MMSG), posterior MSG (PMSG) and PSG. The RNA coverage is given as the log-transformed reads per kilobase of transcript per million mapped reads (RPKM) value, and junction reads are plotted as arcs whose width is proportional to the number of reads aligned to the junction spanning the exons connected by the arc. Genomic coordinates are shown on the X-axis. Quantified mRNA isoforms identified from the MergedSet are shown on the bottom (exons in black, introns as lines with arrowheads)
In addition, we identified 2529 APA events at 2004 genic loci based on the PacBio transcriptome (Table S6). The APA events were further compared with reference genes, which led to the detection of 1629 genes with one poly-A site, 280 genes with two poly-A sites, 66 genes with three poly-A sites, 17 genes with four poly-A sites, four genes with five poly-A sites, and eight genes with more than five poly-A sites (Fig. 3).

Distribution of genes with one or more poly-A sites
Detection of SSRs
As summarized in Table S7, 6596 SSRs from 4148 sequences were identified; 1563 sequences had more than 1 SSR, and 472 were compound SSRs. In addition, the numbers of mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides were 4880, 325, 435, 56, 5, and 2, respectively.
Identification of lncRNAs
To obtain a high-confidence set of lncRNA genes, we identified lncRNAs among the novel transcripts by homologous searches against reference protein databases and combined the results of four prediction methods (CPC/CNCI/CPAT/Pfam). In total, 1403 transcripts were identified as noncoding RNAs (Fig. 4a). Among them, 630 were annotated as lincRNAs (long intergenic noncoding RNAs), 337 were annotated as sense lncRNAs, 147 were annotated as antisense lncRNAs, and 149 were annotated as intronic lncRNAs (Fig. 4b). The densities of lncRNAs, transcripts and fusion events are shown in Fig. 4c. After target gene prediction, a total of 12,663 genes were detected as target genes of lncRNAs, including silk protein structural genes such as fibH, fibL, P25, Ser1 and Ser2 and silk gland factors such as Dimm, Sage, SGF1, SGF2/Awh and FMBP-1 (Table S8). Taken together, these analyses provide some useful clues about the regulation of silk protein synthesis and would reward further study.

Results of lncRNA analysis. a Venn diagram of the lncRNAs predicted by four reference protein databases. b Proportions of four kinds of lncRNAs
Functional annotation of novel full-length (FL) isoforms
Of the 9061 novel transcripts, 2814, 5106, 4009, 5430, 4855, 7296, and 8043 transcripts could be annotated based on the genes in the COG, GO, KEGG, KOG, Swiss-Prot, eggNOG, and NR databases, respectively (Table S9). Annotation of the novel transcripts with the GO database classified 5106 transcripts in three ontologies: cellular component (2430 transcripts), molecular function (4349 transcripts), and biological process (3717 transcripts)(Fig. 5). In the cellular component category, 109 isoforms were annotated to “membrane-enclosed lumen”, which might be associated with silk biosynthesis. A total of 760 isoforms were found to be linked to “macromolecular complex”, which might be related to the mechanism of silk protein assembly. In the molecular function category, 61 isoforms were associated with “electron carrier activity”, “enzyme regulator activity”, “transporter activity”, “nucleic acid binding transcription factor activity”, and “protein binding transcription factor activity”, which might illustrate the processes active in silk gland secretion. In the biological process category, “single-organism process”, “multicellular organismal process” and “rhythmic process” were identified and might reflect the silk synthesis process.

GO annotation of silkworm novel isoform sequences. The x-axis shows three primary categories and 54 subcategories (functional groups). The left side of the Y-axis shows the percentage of the genes, and the right side shows the number of the genes
KEGG was also employed to annotate the novel isoforms. A total of 3521 novel transcripts were associated with 133 unique KEGG pathways (Table S10). Briefly, 53 and two isoforms were associated with the “Insect hormone biosynthesis” and “Steroid biosynthesis” pathways, respectively, which are considered to coordinately regulate the expression of silk protein-coding genes (Bede et al. 2001). Forty isoforms were related to “Hippo signaling pathway—fly”, which has been shown to regulate silk protein synthesis in B. mori (Zeng et al. 2017). Seventy-nine isoforms were predicted to participate in the “Ubiquitin mediated proteolysis” pathway, which indicates a degradation process related to cell death and inflammation and might be helpful in understanding silk gland development (Meier et al. 2015). In addition, some important pathways, including “FoxO signaling pathway”, “Ras signaling pathway” and “MAPK signaling pathway”, were also enriched in novel isoforms and have been reported to be involved in silk protein synthesis (Ma et al. 2011, 2014, 2018). Taken together, this annotation information would help to uncover the biological and metabolic processes involved in silk synthesis.
Identification of TF expressed in silk glands
TFs play important regulatory roles in animal growth and development. In B. mori, some TFs have been reported to play crucial roles in the regulation of silk protein synthesis. We identified 2919 putative TFs belonging to 66 families (Table S11). The top three families identified were “zf-C2H2”, “Homeobox” and “bHLH”. Interestingly, most of the TFs that have previously been shown to regulate the development of silk gland or silk protein synthesis could be found among these 2919 putative TFs, including Antp, Ftz-f1, Sage, Dimm, FMBP-1, SGF3 and SGF1. Five crucial genes, including Antp (BMgn006391), Ftz-f1 (BMgn006393), FMBP-1 (BMgn002810), SGF1 (BMgn005101) and SGF3 (BMgn010868), were selected to generate a phylogenetic tree including genes with the same domains. Genes with similar functions were clustered together in the same clades (Fig. 6). In addition, many TFs involves in developmental signaling pathways, such as 20E and FoxO signaling, were identified, further suggesting the involvement of these pathways in the regulation of silk protein synthesis.

Homeobox evolutionary analysis by the maximum likelihood method. The tree with the highest log likelihood (− 38,011.93) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. This analysis involved 245 amino acid sequences. There were a total of 79 positions in the final dataset
Discussion
In the present study, the FL transcriptome of B. mori silk glands, the unique organ that produces silk proteins, was analyzed using PacBio SMRT. By combining SMRT with SGS data, 11,697 non-redundant transcripts were obtained in total. Meanwhile, 1867 AS events, 1403 lncRNAs, and 784 fusion transcripts were identified, and 2004 genes with 2529 transcripts contained at least one poly-A site. To our knowledge, this is the first study to characterize a B. mori transcriptome using PacBio SMRT, and the results could improve the characterization of the transcriptome of B. mori silk glands.
PacBio sequencing is effective in obtaining reliable FL transcripts. In our study, 51.28% of the PacBio ROIs were FLNC reads with a mean length of 1935 bp, and there was no need to assemble short SGS reads. Combining these long reads with SGS data yielded 28,807 corrected isoforms, and 93.0% were longer than 1 kb. Approximately 5655 isoforms were found to harbor complete ORFs. A total of 6165 genes were detected by SMRT, and their mean length was 2120 bp. PacBio sequencing enriches transcript resources and provides advantages for discovering novel or uncharacterized transcript isoforms and genes. We identified 6896 novel isoforms from known genes and 2165 novel isoforms from novel genes in the reference genome based on SMRT data. These data not only enrich the transcript information of the draft genome sequence but also support functional studies of important genes in further research.
Previous transcriptome analyses in B. mori have mainly relied on SGS technology, which is often unable to accurately capture or assemble FL transcripts. With PacBio SMRT technology, RNA fragmentation is not required, and intact transcript sequence information is provided, avoiding assembly. In our work, transcript completeness rose to 94.8%, and the N50 rose to 2439 bp with remarkably fewer ambiguous sites (N bases). PacBio sequencing also promotes the identification of AS events. We detected 1867 AS events in the FLNC reads, most of which were IR events; these results greatly enriched the transcript information in the draft version of the B. mori genome. In contrast, the majority of the AS events in the reference genome were AA splices.
In this study, we found 784 fusion transcripts, and fusion events were more likely to occur interchromosomally than intrachromosomally. The chimeric fusion events in B. mori enhance the complexity of the silkworm transcriptome. LncRNAs, a hotspot of molecular biology, are thought to be important regulators, but their functions are not yet completely understood. We identified 1403 lncRNAs with a mean length of 1963.96 bp, and most were lincRNAs. In addition, we found that 2529 isoforms at 2004 genic loci had at least one poly-A site. These results provide useful information for future analysis of the relationship of APA to the functions of genes related to silk gland or silk protein synthesis.
In this study, for the first time, we performed PacBio SMRT sequencing of the FL transcriptome of B. mori silk glands. The obtained transcriptome will greatly revise and expand the available B. mori transcripts and facilitate further studies of the genes involved in the regulation of silk protein synthesis.
References
Abdel-Ghany SE, Hamilton M, Jacobi JL, Ngam P, Devitt N, Schilkey F, Ben-Hur A, Reddy AS (2016) A survey of the sorghum transcriptome using single-molecule long reads. Nat Commun 7:11706
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Bede JC, Teal PE, Goodman WG, Tobe SS (2001) Biosynthetic pathway of insect juvenile hormone III in cell suspension cultures of the sedge Cyperusiria. Plant Physiol 127(2):584–593
Beier S, Thiel T, Münch T, Scholz U, Mascher M (2017) MISA-web: a web server for microsatellite prediction. Bioinformatics 33(16):2583–2585
Chang H, Cheng T, Wu Y, Hu W, Long R, Liu C, Zhao P, Xia Q (2015) Transcriptomic analysis of the anterior silk gland in the domestic silkworm (Bombyx mori)—insight into the mechanism of silk formation and spinning. PLoS ONE 10(9):e0139424
Chen J, Tang X, Ren C, Wei B, Wu Y, Wu Q, Pei J (2018) Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom 19(1):548
Cheng B, Furtado A, Henry RJ (2017) Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 6(11):1–13
Couble P, Michaille JJ, Garel A, Couble ML, Prudhomme JC (1987) Developmental switches of sericin mRNA splicing in individual cells of Bombyx mori silk gland. Dev Biol 124(2):431–440
Cui Y, Zhu Y, Lin Y, Chen L, Feng Q, Wang W, Xiang H (2018) New insight into the mechanism underlying the silk gland biological process by knocking out fibroin heavy chain in the silkworm. BMC Genom 19(1):215
Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, Xia Q (2010) SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res 38(Database issue):D453–D456
Fang SM, Hu BL, Zhou QZ, Yu QY, Zhang Z (2015) Comparative analysis of the silk gland transcriptomes between the domestic and wild silkworms. BMC Genom 16:60
Foissac S, Sammeth M (2007) ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res 35(Web Server issue):W297–W299
Garel A, Deleage G, Prudhomme JC (1997) Structure and organization of the Bombyx mori sericin 1 gene and of the sericins 1 deduced from the sequence of the Ser 1B cDNA. Insect Biochem Mol Biol 27(5):469–477
Hu W, Chen Y, Lin Y, Xia Q (2019) Developmental and transcriptomic features characterize defects of silk gland growth and silk production in silkworm naked pupa mutant. Insect Biochem Mol Biol 111:103175
Katz Y, Wang ET, Airoldi EM, Burge CB (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7(12):1009–1015
Katz Y, Wang ET, Silterra J, Schwartz S, Wong B, Thorvaldsdóttir H, Robinson JT, Mesirov JP, Airoldi EM, Burge CB (2015) Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31(14):2400–2402
Korlach J, Bjornson KP, Chaudhuri BP, Cicero RL, Flusberg BA, Gray JJ, Holden D, Saxena R, Wegener J, Turner SW (2010) Real-time DNA sequencing from single polymerase molecules. Methods Enzymol 472:431–455
Kuo RI, Tseng E, Eory L, Paton IR, Archibald AL, Burt DW (2017) Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genom 18(1):323
Li G, Li Y, Chen G, He J, Han Y, Wang X, Kaplan DL (2015) Silk-based biomaterials in biomedical textiles and fiber-based implants. Adv Healthc Mater 4(8):1134–1151
Ma L, Xu H, Zhu J, Ma S, Liu Y, Jiang RJ, Xia Q, Li S (2011) Ras1(CA) overexpression in the posterior silk gland improves silk yield. Cell Res 21(6):934–943
Ma L, Ma Q, Li X, Cheng L, Li K, Li S (2014) Transcriptomic analysis of differentially expressed genes in the Ras1(CA)-overexpressed and wildtype posterior silk glands. BMC Genom 15:182
Ma L, Li K, Guo Y, Sun X, Deng H, Li K, Feng Q, Li S (2018) Ras-Raf-MAPK signaling promotes nuclear localization of FOXA transcription factor SGF1 via Ser91 phosphorylation. Biochim Biophys Acta Mol Cell Res 1865(4):560–571
Meier P, Morris O, Broemer M (2015) Ubiquitin-mediated regulation of cell death, inflammation, and defense of homeostasis. Curr Top Dev Biol 114:209–239
Michael TP, VanBuren R (2015) Progress, challenges and the future of crop genomes. Curr Opin Plant Biol 24:71–81
Obara T, Suzuki Y (1988) Temporal and spatial control of silk gene transcription analyzed by nuclear run-on assays. Dev Biol 127(2):384–391
Omenetto FG, Kaplan DL (2010) New opportunities for an ancient material. Science 329(5991):528–531
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genom Proteom Bioinform 13(5):278–289
Royer C, Briolay J, Garel A, Brouilly P, Sasanuma S, Sasanuma M, Shimomura M, Keime C, Gandrillon O, Huang Y, Chavancy G, Mita K, Couble P (2011) Novel genes differentially expressed between posterior and median silk gland identified by SAGE-aided transcriptome analysis. Insect Biochem Mol Biol 41(2):118–124
Sharon D, Tilgner H, Grubert F, Snyder M (2013) A single-molecule long-read survey of the human transcriptome. Nat Biotechnol 31(11):1009–1014
Shen Y, Zhou Z, Wang Z, Li W, Fang C, Wu M, Ma Y, Liu T, Kong LA, Peng DL, Tian Z (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26(3):996–1008
Shi R, Ma S, He T, Peng J, Zhang T, Chen X, Wang X, Chang J, Xia Q, Zhao P (2019) Deep insight into the transcriptome of the single silk gland of Bombyx mori. Int J Mol Sci 20(10):E2491
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212
Song J, Che J, You Z, Ye L, Li J, Zhang Y, Qian Q, Zhong B (2016) Phosphoproteomic analysis of the posterior silk gland of Bombyx mori provides novel insight into phosphorylation regulating the silk production. J Proteom 148:194–201
Suetsugu Y, Futahashi R, Kanamori H, Kadono-Okuda K, Sasanuma S, Narukawa J, Ajimura M, Jouraku A, Namiki N, Shimomura M, Sezutsu H, Osanai-Futahashi M, Suzuki MG, Daimon T, Shinoda T, Taniai K, Asaoka K, Niwa R, Kawaoka S, Katsuma S, Tamura T, Noda H, Kasahara M, Sugano S, Suzuki Y, Fujiwara H, Kataoka H, Arunkumar KP, Tomar A, Nagaraju J, Goldsmith MR, Feng Q, Xia Q, Yamamoto K, Shimada T, Mita K (2013) Large scale full-length cDNA sequencing reveals a unique genomic landscape in a lepidopteran model insect, Bombyx mori. G3 (Bethesda) 3(9):1481–1492
Takasu Y, Hata T, Uchino K, Zhang Q (2010) Identification of Ser2 proteins as major sericin components in the non-cocoon silk of Bombyx mori. Insect Biochem Mol Biol 40(4):339–344
Tomita M (2011) Transgenic silkworms that weave recombinant proteins into silk cocoons. Biotechnol Lett 33(4):645–654
Wang B, Tseng E, Regulski M, Clark TA, Hon T, Jiao Y, Lu Z, Olson A, Stein JC, Ware D (2016a) Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun 7:11708
Wang S, You Z, Feng M, Che J, Zhang Y, Qian Q, Komatsu S, Zhong B (2016b) Analyses of the molecular mechanisms associated with silk production in silkworm by iTRAQ-based proteomics and RNA-sequencing-based transcriptomics. J Proteome Res 15(1):15–28
Wen M, Ng JHJ, Zhu F, Chionh YT, Chia WN, Mendenhall IH, Lee BP, Irving AT, Wang LF (2018) Exploring the genome and transcriptome of the cave nectar bat Eonycterisspelaea with PacBio long-read sequencing. Gigascience 7(10):giy116
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21(9):1859–1875
Xia Q, Cheng D, Duan J, Wang G, Cheng T, Zha X, Liu C, Zhao P, Dai F, Zhang Z, He N, Zhang L, Xiang Z (2008) Microarray-based gene expression profiles in multiple tissues of the domesticated silkworm, Bombyx mori. Genome Biol 8(8):R162
Yi S, Zhou X, Li J, Zhang M, Luo S (2018) Full-length transcriptome of Misgurnus anguillicaudatus provides insights into evolution of genus Misgurnus. Sci Rep 8(1):11699
Zeng W, Liu R, Zhang T, Zuo W, Ou Y, Tang Y, Xu H (2017) BmYki is transcribed into four functional splicing isoforms in the silk glands of the silkworm Bombyx mori. Gene 646:39–46
Zhong BX, Yu YP, Xu YS, Yu H, Lu XM, Miao YG, Yang J, Xu H, Hu SN, Lou CF (2005) Analysis of ESTs and gene expression patterns of the posterior silk gland in the fifth instar larvae of silkworm Bombyx mori L. Sci China (C) 48(1):25–33
Acknowledgements
This work was supported by a grant (31872291) from the National Natural Science Foundation of China and a grant (cstc2017jcyjBX0041) from the Chongqing Research Program of Basic Research and Frontier Technology. American Journal Experts performed English language editing on this manuscript.
Author information
Authors and Affiliations
Contributions
All authors conceived and designed the experiments. TC, QS, YM and WZ performed the experiments and analyzed the data. LH, QS and HX wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors
Additional information
Communicated by S. Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2020_1691_MOESM1_ESM.tif
Supplementary file1 (TIF 2897 kb) Bioinformatics analysis process. (A) PacBio data processing pipeline. (B) Transcriptome analysis pipeline
438_2020_1691_MOESM2_ESM.tif
Supplementary file2 (TIF 410 kb) AS events in major silk gland TFs. Sashimi plot of ES and truncation events in the ASG, MMSG, PMSG and PSG. The RNA coverage is given as the log-transformed RPKM value, and junction reads are plotted as arcs whose width is proportional to the number of reads aligned to the junction spanning the exons connected by the arc. Genomic coordinates are shown on the x-axis. Quantified mRNA isoforms identified from the MergedSet are shown on bottom (exons in black, introns as lines with arrowheads)
Rights and permissions
About this article

Cite this article
Chen, T., Sun, Q., Ma, Y. et al. A transcriptome atlas of silkworm silk glands revealed by PacBio single-molecule long-read sequencing. Mol Genet Genomics 295, 1227–1237 (2020). https://doi.org/10.1007/s00438-020-01691-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-020-01691-9