
Abstract
The xylA gene from a marine bacterium, Vibrio sp. strain XY-214, encoding d-xylose isomerase (XylA) was cloned and expressed in Escherichia coli. The xylA gene consisted of 1,320-bp nucleotides encoding a protein of 439 amino acids with a predicted molecular weight of 49,264. XylA was classified into group II xylose isomerases. The native XylA was estimated to be a homotetramer with a molecular mass of 190 kDa. The purified recombinant XylA exhibited maximal activity at 60°C and pH 7.5. Its apparent K m values for d-xylose and d-glucose were 7.93 and 187 mM, respectively. Furthermore, we carried out d-xylulose production from β-1,3-xylan, a major cell wall polysaccharide component of the killer alga Caulerpa taxifolia. The synergistic action of β-1,3-xylanase (TxyA) and β-1,3-xylosidase (XloA) from Vibrio sp. strain XY-214 enabled efficient saccharification of β-1,3-xylan to d-xylose. d-Xylose was then converted to d-xylulose by using XylA from the strain XY-214. The conversion rate of d-xylose to d-xylulose by XylA was found to be approximately 40% in the presence of 4 mM sodium tetraborate after 2 h of incubation. These results demonstrated that TxyA, XloA, and XylA from Vibrio sp. strain XY-214 are useful tools for d-xylulose production from β-1,3-xylan. Because d-xylulose can be used as a source for ethanol fermentation by yeast Saccharomyces cerevisiae, the present study will provide a basis for ethanol production from β-1,3-xylan.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, growing attention has been devoted to the conversion of biomass into the fuel ethanol, and significant advances have been made toward the technology of saccharification of carbohydrate polymers and ethanol fermentation (Lin and Tanaka 2006). In most cases, terrestrial plants, such as agricultural and forestry residues, are used for ethanol production because these contain large amounts of potentially fermentable sugars in the form of starch, cellulose, and β-1,4-xylan. In contrast, studies involved in ethanol production from algal biomass are very few.
The mutant Caulerpa taxifolia called as killer green alga was the first macrophyte invasion that drew widespread public attention. Its first breakout is attributed to an aquarium in Monaco in 1984 (Jousson et al. 1998; Olsen et al. 1998; Meusnier et al. 2001), and the invasive alga has now spread to the coasts of six countries bordering the Mediterranean Sea, i.e., Croatia, France, Italy, Monaco, Spain, and Tunisia (Meinesz and Hesse 1991; Meinesz et al. 2001). Furthermore, C. taxifolia was discovered for the first time in Californian coast in 2000 (Dalton 2000; Kaiser 2000). Additionally, new introduced populations of C. taxifolia were recognized in Australia (New South Wales; Schaffelke et al. 2002). Although C. taxifolia destroys the ecosystems of the sea environment through competition with native species, attempts at management of the invasive algae in the Mediterranean Sea remain rare.
β-1,3-Xylan is a major cell wall polysaccharide component of Caulerpa spp. (Iriki et al. 1960). It is also found in the cell walls of other green algae, such as Bryopsis and Udotea spp., and red algae (Porphyra and Bangia spp.; Iriki et al. 1960; McDowell 1967). Because β-1,3-xylan is a homopolysaccharide composed of β-1,3-linked d-xylose residues, it can be used as a source for ethanol fermentation. Similar to other biomass materials such as starch, cellulose, and β-1,4-xylan, the saccharification process using glycoside hydrolases is required for ethanol production from β-1,3-xylan. However, there are a few studies about β-1,3-xylan-degrading enzymes (Chen et al. 1986; Aoki et al. 1988; Yamaura et al. 1990; Araki et al. 1998, 1999).
Vibrio sp. strain XY-214 is a marine bacterium that can grow on β-1,3-xylan as a sole carbon source (Araki et al. 1999). To date, we have cloned and characterized the genes encoding a β-1,3-xylanase (TxyA; 1,3-β-d-xylan xylanohydrolase; EC 3.2.1.32; Araki et al. 2000) and a β-1,3-xylosidase (XloA; Umemoto et al. 2008) from the bacterium. The synergistic action of these two types of enzymes enables the complete degradation of β-1,3-xylan to d-xylose. Although yeast Saccharomyces cerevisiae is the most effective ethanol-producing organism, it cannot ferment d-xylose as a carbon source. In contrast, the yeast can ferment d-xylulose, a keto isomer of d-xylose (Gong et al. 1981). Therefore, d-xylose released from β-1,3-xylan must be converted to d-xylulose before ethanol fermentation.
d-Xylose isomerase (XIs; EC 5.3.1.5) is an intracellular enzyme which catalyzes the reversible isomerization of d-xylose to d-xylulose in the first step of the d-xylose metabolism following the pentose phosphate pathway. The enzyme can also isomerize d-glucose to d-fructose. However, K m is the lowest and V max is the greatest when d-xylose is the substrate (Meng et al. 1991; Rangarajan and Hartley 1992). The XIs can be classified into two groups based on their amino acid sequences (group I enzymes are shorter by approximately 50 residues at the N terminus; Park and Batt 2004).
Fortunately, our previous study revealed that a truncated gene (xylA), which showed high sequence similarities to several bacterial XIs, is located in the upstream region of the β-1,3-xylosidase gene (xloA) from Vibrio sp. strain XY-214 (Umemoto et al. 2008). In the present study, therefore, we first cloned the complete xylA gene encoding a d-xylose isomerase from the genomic DNA of the strain XY-214 and characterized a recombinant d-xylose isomerase (rXylA) expressed in a transformed Escherichia coli. Second, we tried to produce d-xylulose from β-1,3-xylan by using three kinds of recombinant enzymes (rTxyA, rXloA, and rXylA). Our purpose is to provide information regarding the utilization of a killer alga, C. taxifolia, by the establishment of a basis for ethanol production from β-1,3-xylan.
Materials and Methods
Materials
β-1,3-Xylan was prepared from a green alga, Caulerpa racemosa var. laetevirens, by the method of Iriki et al. (1960). d-Xylulose was purchased from Sigma Chemical (St. Louis, MO, USA). d-Xylose, d-glucose, and the other chemicals were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Bacterial Strains, Plasmids, and Culture Conditions
Vibrio sp. strain XY-214, isolated from sea mud at Ise bay in 1990, was grown in peptone medium (0.5% peptone, 0.1% yeast extract, 3% NaCl, 0.05% MgSO4, 0.2% K2HPO4, 0.04% KH2PO4; pH 7.6) containing 0.3% β-1,3-xylan and used as the source of chromosomal DNA. E. coli XL1-Blue and pBluescript II KS(−) plasmids served as cloning hosts and vectors (Stratagene, La Jolla, CA, USA), respectively. E. coli BL21 (DE3; Novagen, Madison, WI, USA) was used as the host for a derivative of pET22b(+) (Novagen) in the production of a recombinant protein. All E. coli strains were grown in Luria–Bertani (LB) medium supplemented with ampicillin (50 μg ml−1). For agar medium, LB medium was solidified with 1.5% agar.
Cloning of the xylA Gene
Previously, we have reported the cloning of a novel gene encoding β-1,3-xylosidase from Vibrio sp. strain XY-214. It has been revealed that a 4,206-bp DNA fragment, which was obtained by Southern hybridization analysis with a partial xloA fragment as the probe, contains the complete xloA gene and three possible open reading frames (ORFs; Umemoto et al. 2008). Sequence analysis of the DNA fragment revealed that an incomplete ORF encoding a putative XylA was located in the upstream region of the xloA gene. On the other hand, two complete ORFs named gatA and alrA are located in the downstream of the xloA gene. The gatA and alrA gene products showed similarity to a galactoside acetyltransferase (GAT) and an aldehyde reductase (AlrA), respectively. On the basis of the sequence of a partial xylA gene, one specific primer set (pxylA-F, 5′-CTAGAGAACGAGATCAAAGT-3′, and pxylA-R, 5′-CTCTAGCGATAGTGCCATAG-3′) was designed. The PCR product (330 bp) amplified from the plasmid xloA/pBluescript (XbaI-SpeI; Umemoto et al. 2008) with the primer set was used as the probe for cloning of the xylA gene after it was labeled with alkaline phosphatase (GE Healthcare, Buckinghamshire, England) according to the manufacturer’s instructions.
To obtain the complete xylA gene, Southern hybridization with the probe was carried out. Digestion of Vibrio sp. strain XY-214 genomic DNA with both SacI and EcoRI gave a 4.2-kbp fragment that was hybridized with the AlkPhos-labeled probe. The DNA fragments corresponding to 4.2-kbp were excised from the gel and purified with a Wizard gel and PCR clean-up system (Promega Co., Madison, WI, USA). These were ligated into the SacI and EcoRI sites of pBluescript II KS(−) (Stratagene), and the recombinant plasmids were introduced into E. coli XL1-Blue (Stratagene). One of the 123 colonies was selected as positive by colony hybridization with the AlkPhos-labeled probe. The cloned plasmid was named xylA/pBluescript (SacI-EcoRI).
DNA Sequencing and Bioinformatics Approaches
Nucleotide sequence analysis of the DNA fragment inserted into the vector was carried out on a Beckman CEQ2000XL sequencer (Beckman Coulter, Fullerton, CA, USA) using a GenomeLabTM DTCS-Quick Start Kit (Beckman Coulter). Oligonucleotide primers designed on the basis of known sequences were also used in DNA sequencing. The nucleotide sequence data were analyzed using GENETYX-WIN computer software (Genetyx Corporation, Tokyo, Japan). Similarity searches were performed using the basic local alignment search tool algorithm at the National Center for Biotechnology Information server (Altschul et al. 1990). The molecular mass of a gene product was estimated by using the peptide mass tool at the ExPASy server of the Swiss Institute of Bioinformatics (Gasteiger et al. 2003).
Expression and Purification of rXylA
For the production of the rXylA in E. coli, the xylA gene was subcloned into a pET22b(+) vector (Novagen) as follows: The full-length xylA gene was amplified from xylA/pBluescript (SacI-EcoRI) as the template by PCR using the primers 5′-AGGGCCCATATGACTGAATTTTTCAAA-3′ and 5′-AGCTCGAGCTTGTAGATGAAGCCATTTA-3′, which contained artificial NdeI and XhoI sites (underlined), respectively. The PCR product was digested with NdeI and XhoI and ligated into a pET22b(+) vector linearized with the same enzymes to construct the xylA/pET22b. This plasmid provides the recombinant XylA with a six-His tag fused at its C terminus. The xylA/pET22b was transformed into E. coli BL21 (DE3)-competent cells (Novagen) and used for the production of rXylA. The absence of undesired mutations in the amplified DNA fragment was verified by DNA sequencing.
E. coli BL21 (DE3) transformants carrying xylA/pET22b were cultivated at 37°C in 800 ml of LB medium in the presence of ampicillin (50 μg ml−1). When the absorbance of the culture reached around 0.6 at 600 nm, the temperature was changed to 25°C and isopropyl-β-d-galactopyranoside was added to the culture to give a final concentration of 1 mM for the induction of gene expression. After an additional incubation of 20 h at 25°C, the cells harvested by centrifugation were suspended in binding buffer (20 mM sodium phosphate, 0.5 M NaCl, and 10 mM imidazole; pH 7.4) and disrupted on ice by sonication. The supernatant of the cell lysate was collected by centrifugation and fractionated by a HiTrap chelating HP column (GE Healthcare) precharged with Ni2+. The recombinant protein was eluted with a linear gradient of imidazole (10–500 mM). The purified rXylA was dialyzed against 50 mM sodium phosphate buffer (pH 7.5) and stored at 4°C until use. Subunit molecular masses and enzyme purity were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 12% polyacrylamide gels (Laemmli 1970). The protein concentration was measured by the method of Lowry et al. (1951) with bovine serum albumin (Sigma Chemicals Co.) as a standard.
Enzyme Assays
d-Xylose isomerase activity was determined by measuring the accumulation of d-xylulose. Unless otherwise indicated, the standard reaction mixture contained 5 mM d-xylose, 5 mM MgSO4, the enzyme solution at a suitable dilution, and 50 mM sodium phosphate buffer (pH 7.5) to bring the final volume to 400 μl. The reaction mixture was incubated at 40°C for 10 min followed by cooling the tubes on ice to stop the reaction. The d-xylulose formed was quantified by the cysteine–carbazole–sulfuric acid method (Dische and Borenfreund 1951), and the absorbance was measured at 540 nm. One unit of isomerase activity was defined as the amount of enzyme that produced 1 μmol of keto-sugar per minute under the assay conditions.
Characterization of rXylA
The effects of temperature on the activity of rXylA were determined for a range between 0°C to 80°C. With the exception of temperature, the assay conditions were the same as those described for the standard method. For the temperature stability measurements, the enzymes were preincubated at various temperatures at pH 7.5 for 20 min, and the residual activity was measured using the standard method. The effects of pH on the activity and stability of rXylA were examined at various pHs (3.0–12.0) in Britton and Robinson’s universal buffer (40 mM phosphoric acid, 40 mM boric acid, and 40 mM acetic acid). The pH activities were assayed under the conditions described for the standard method. For the pH stability measurements, the enzymes were preincubated at various pHs at 4°C for 12 h, and the residual activity was then measured using the standard method.
To determine the kinetic parameters, enzyme assays were performed in 50 mM sodium phosphate buffer (pH 7.5) containing 5 mM MgSO4 and 1–64 mM d-xylose (25–300 mM for d-glucose). The reaction mixtures were incubated for 10 min at 40°C, and then reactions were stopped by cooling the tubes in ice. The d-xylulose and d-fructose formed were quantified by cysteine–carbazole–sulfuric acid method, and the absorbance at 540 and 560 nm were measured, respectively. The apparent K m (millimolars per liter) and V max (units per milligram protein) were calculated from Lineweaver–Burk plots.
For the determination of native molecular mass, samples were applied to a Superdex 200 HR 10/30 column (GE Healthcare) equilibrated with 50 mM sodium phosphate buffer (pH 7.5) containing 0.15 M NaCl. Molecular mass standards (GE Healthcare) included ferritin (440 kDa), aldolase (158 kDa), ovalbumin (43 kDa), and chymotrypsinogen A (25 kDa).
Effect of Metal Ions on rXylA Activity
Metal ions were removed from the purified rXylA by treatment with 10 mM EDTA at 4°C for 1 h, followed by overnight dialysis against 50 mM sodium phosphate buffer (pH 7.5) at 4°C with several changes of buffer solution. The effects of various metal ions were determined by adding 5 mM MgSO4 CoCl2, MnCl2, CaCl2, ZnSO4, FeSO4, or CuSO4 to the dialyzed enzyme and assaying d-xylose isomerase activity under standard conditions without 5 mM MgSO4.
Saccharification of β-1,3-Xylan to d-Xylose
β-1,3-Xylanase (rTxyA) and β-1,3-xylosidase (rXloA) were used for the saccharification of β-1,3-xylan. Both enzymes were expressed in E. coli transformants and purified as described previously (Araki et al. 2000; Umemoto et al. 2008). Then, 0.5% β-1,3-xylan in 50 mM sodium phosphate buffer (pH 7.5) were placed in Eppendorf tubes in the presence of 2.5 U ml−1 rTxyA and 0.15 U ml−1 rXloA either alone or in combination (total volume 0.3 ml). The reaction was carried out at 37°C for 12 h. After the hydrolysis reactions were stopped by boiling the tubes for 5 min, the supernatants separated by centrifugation were used for analysis of hydrolysis products. The amount of reducing sugar released from β-1,3-xylan was measured by the Somogyi–Nelson method (Somogyi 1952). The hydrolysis products were detected by thin-layer chromatography (TLC) on a silica gel 60 plastic sheet (Merck, Darmstadt, Germany) with a solvent of n-butanol–acetic acid–water (10:5:1, v/v). Oligosaccharides were visualized by spraying the plate with diphenylamine–aniline–phosphate reagent (Bailey and Bourne 1960).
d-Xylulose Production from d-Xylose
The reaction mixture containing 50 mM sodium phosphate buffer (pH 7.5), 1.5% d-xylose, 10 mM MgSO4, 4 mM sodium tetraborate, and 0.25 U rXylA in Eppendorf tubes (total volume 1 ml) were incubated at 40°C for 2 h. Sodium tetraborate has been reported to shift the equilibrium constant between d-xylose and d-xylulose to favor the formation of d-xylulose (Hsiao et al. 1982). The borate ion binds more tightly to d-xylulose than d-xylose, effectively reducing the free d-xylulose concentration, and thus leads to increased d-xylose isomerization. The binding ability of borate ion to d-xylulose is pH dependent, with higher pH (6.0–7.5) favoring binding (Hsiao et al. 1982). The borate ion dissociates from d-xylulose in lower pH. To analyze the conversion rates of d-xylose to d-xylulose, the reaction mixtures were boiled for 2 min to inactivate any residual d-xylose isomerase activity. The samples were then centrifuged and the pH of the supernatant was adjusted to 5.0 by 1 M acetic acid. These preparations were used for analyses of the reaction products. Sugar composition was analyzed by high-performance liquid chromatography (HPLC) using a refractive index (RI) detector (Shodex RI-72; Showa Denko, Tokyo, Japan) together with a Shodex Asahipak NH2P-50 4E column (4.6 mm i.d. × 250 mm; Showa Denko). Elution was performed at a flow rate of 1.0 ml min−1 using acetonitrile–water (75:25, v/v) as the mobile phase. The column temperature was maintained at 40°C.
Nucleotide Sequence Accession Number
The nucleotide sequence of the xylA gene described here has been submitted to the DDBJ, EMBL, and GenBank databases and assigned the accession no. AB519447.
Results
Cloning and Nucleotide Sequence Analysis of the xylA Gene
Nucleotide sequence analysis revealed that the plasmid xylA/pBluescript (SacI-EcoRI) contained 4,242-bp DNA fragment, including the complete xylA gene and the other two genes named xylR and xylT. The xylA gene was located in the upstream region of the xloA gene in the same direction (Fig. 1). There are only 80 nucleotides between the TAA termination codon of the xylA gene and the ATG initiation codon of the xloA gene. The xylA gene consisted of 1,320-bp nucleotides encoding a protein comprising 439 amino acids with a calculated molecular mass of 49,264 Da. The sequences TTGATG and TAAAAT with 15-bp spacing, having a certain homology to the −35 and −10 promoter consensus sequences of E. coli, were identified upstream of the coding region (Rosenberg and Court 1979). A putative ribosome-binding site sequence (GGAA) was located within a few bases upstream of the ATG start codon. A possible transcription terminator that consisted of a 36-bp palindrome sequence corresponding to an mRNA hairpin loop, followed by a T-rich portion, was found downstream of the TAA termination codon (Sugimoto et al. 1995). The xylT and xylR genes were located in the upstream region of the xylA gene in the opposite transcriptional directions, while the C-terminal region of the xylR gene was truncated. The N-terminal region (residues 4 to 137) of the xylR gene product showed similarities to xylose operon transcriptional regulators from Klebsiella pneumoniae 342 (31% identity, GenBank accession no. CP000964; residues 18 to 143) and Yersinia pestis KIM (30% identity, AALD02000013; residues 18 to 149). The xylT gene consisted of a 1,389-bp nucleotide sequence encoding a protein comprising 462 amino acids with a predicted molecular mass of 50,483 Da. The deduced amino acid sequence of the xylT gene product exhibits similarities to several sugar transporters, such as the Na+/melibiose symporter from Saccharophagus degradans 2–40 (38% identity, CP000282; residues 1 to 538) and Na+/xyloside symporter from Lactobacillus brevis ATCC 367 (31% identity, CP000416; residues 1 to 466).

Genomic organization of β-1,3-xylan utilization gene cluster in Vibrio sp. strain XY214. pTxyA, pXloA, and pXloA are the DNA fragments cloned for the txyA, xloA, and xylA genes, respectively. The xylR and xbpA genes are truncated. Hairpin-like marks indicate putative transcription terminators
Identification of the β-1,3-Xylan Utilization Gene Cluster
As shown in Fig. 1, the 7,267-bp DNA fragment, which consists of six different genes organized in the order xylR, xylT, xylA, xloA, gatA, and alrA, was generated from the 4,206-bp and 4,242-bp DNA fragments obtained for the cloning of the xloA and xylA genes, respectively. In addition, we found that the 3′ region of alrA gene, which encodes an aldehyde reductase (Umemoto and Araki 2008), was overlapped with the 5′ region of the 4,409-bp DNA fragment obtained for the cloning of a β-1,3-xylanase gene (txyA) from Vibrio sp. strain XY-214 (Araki et al. 2000). These results indicate that Vibrio sp. strain XY-214 has a gene cluster involved in the β-1,3-xylan degradation and d-xylose metabolism. The gene cluster obtained is 11,101-bp long and contains eight genes. There is an incomplete ORF that encodes a protein (XbpA) of unknown function in the upstream region of the txyA gene. However, the N-terminal sequence of the XbpA showed high sequence similarity with a family 31 carbohydrate-binding module (CBM31) of TxyA (Okazaki et al. 2002; Kiyohara et al. 2005). Carbohydrate-binding modules (CBMs) are non-catalytic protein modules found in many carbohydrate-degrading enzymes, and they function as recognition modules that convey the catalytic modules of these enzymes to the target substrate (Boraston et al. 2004). The CBM31 has a specific binding ability to β-1,3-xylan (Okazaki et al. 2002). Therefore, it is possible that XbpA is also involved in the β-1,3-xylan metabolism.
Similarity of the Deduced Amino Acid Sequence Encoded by the xylA Gene
The deduced amino acid sequence of the XylA from Vibrio sp. strain XY-214 was scanned with BLAST, and the protein showed similarities to other bacterial XIs. XylA exhibited the high sequence identities to the group II XIs (identity percentages are in parentheses) from E. coli (72%, GenBank accession no. AE014075), K. pneumoniae 342 (70%, CP000964), Thermotoga maritima MSB8 (52%, AE000512), Bacillus licheniformis DSM 13 (51%, AE017333), and Lactococcus lactis (46%, AF092042). In contrast, XylA showed low sequence identities to the group I XIs from Streptomyces rubiginosus (28%, M73789), Actinoplanes missouriensis (25%, X16042), and Arthrobacter sp. B-5-MG-1 (25%, AY937238). This suggested that XylA of Vibrio sp. strain XY-214 should be classified into the group II enzymes. Sequence alignment of XylA with other XIs revealed that all of the active-site residues (W49, F60, H101, D104, F145, W188, E232, K234, E237, N266, E268, H271, and D296) and residues mediating subunit interactions (D58, R191, L254, and A275; Park and Batt 2004) were conserved in XylA (Fig. 2).

Sequence alignment of the amino acid sequences of several d-xylose isomerases. From top to bottom: B. licheniformis DSM 13 (B.l.), E. coli (E.c), K. pneumoniae 342 (K.p.), L. lactis (L.l.), T. maritima MSB8 (T.m.), and Vibrio sp. strain XY-214 (V.s.). Conserved amino acid residues are highlighted. The conserved active-site residues and residues mediating subunit interactions are indicated by closed and open circles, respectively
Biochemical Properties of rXylA
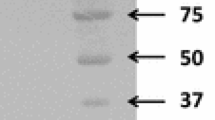
The purified rXylA was shown to be homogeneous by the detection of a single protein band on SDS-PAGE (Fig. 3, lane 3). Its relative molecular mass was estimated to be approximately 48 kDa. However, the relative molecular mass of the native enzyme was estimated to be 190 kDa by gel filtration chromatography on a Superdex 200 HR 10/30 column (data not shown). According to the nucleotide sequence, the relative molecular mass of the XylA monomer should be 49,264 Da. These results suggest that XylA is a homotetramer, which is the most common oligomer form of d-xylose isomerases (Hess et al. 1998).

SDS-PAGE of purified rTxyA, rXloA, and rXylA. Lane M molecular weight markers, lane 1 rTxyA, lane 2 rXloA, lane 3 rXylA
The optimum pH of the rXylA was observed to be around 7.5 when the enzyme activity was assayed with d-xylose at 40°C for 20 min at various pHs (3.0–12.0) in Britton and Robinson’s universal buffer (Fig. 4a). The enzyme was stable at pH 6.0–11.0, retaining more than 80% of the maximal activity after preincubation in the buffer at various pHs at 4°C for 12 h (data not shown). The enzyme was optimally active at around 60°C (Fig. 4b) and stable up to 50°C for 20 min. However, only 34% of the maximal activity was observed after preincubation at 60°C for 20 min, and no activity was observed after preincubation at 70°C for 20 min. The kinetic parameters were determined for d-xylose and d-glucose as substrates from Lineweaver–Burk plots. The K m, V max, K cat, and K cat/K m for d-xylose were 7.93 mM, 14.3 U mg−1, 47.0 s−1, and 5.93 × 103 M−1 s−1, respectively. On the other hand, the K m and V max, K cat, and K cat/K m for d-glucose were 187 mM, 1.38 U mg−1, 4.54 s−1 and 2.40 × 10 M−1 s−1, respectively.

Effects of pH (a) and temperature (b) on d-xylose isomerase activity of the purified rXylA. Each value represents the averages of at least duplicate measurements
Typically, two divalent cations (Mg2+, Co2+, or Mn2+) per monomer are required for catalytic activity and stability of d-xylose isomerases (Carrell et al. 1989). However, the enzymes from different organisms require different metals for optimum activity. As shown in Table 1, rXylA requires the addition of Mg2+ for optimum activity, while the addition of Co2+ or Mn2+ gave lower activities. Addition of Ca2+, Zn2+, Fe2+, or Cu2+ was not effective. No activity was observed in the absence of metal ions.
Saccharification of β-1,3-Xylan
Saccharification of β-1,3-xylan was carried out by using the purified rTxyA (Fig. 3, lane 1) and rXloA (Fig. 3, lane 2) at 37°C and pH 7.5. rTxyA hydrolyzes β-1,3-xylan to β-1,3-xylooligosaccharides, and then rXloA hydrolyzes β-1,3-xylooligosaccharides to d-xylose. Figure 5a shows the time course of the hydrolysis of β-1,3-xylan. A large amount of reducing sugar was released (2.62 g l−1) by the co-action of rTxyA and rXloA against β-1,3-xylan, whereas the action of rTxyA alone gave poor hydrolysis (1.39 g l−1) in a 12 h of hydrolysis. To examine the hydrolysis products released from β-1,3-xylan, TLC analysis was carried out. Samples subjected to a 12 h of hydrolysis were used for TLC analysis. As shown in Fig. 5b, d-xylose was released from β-1,3-xylan as the major hydrolysis product by the co-action of rTxyA and rXloA, whereas the action of rTxyA alone released predominantly β-1,3-xylobiose with d-xylose and β-1,3-xylotriose as minor products. No hydrolysis products were observed by the action of rXloA alone.

The time course of hydrolysis of β-1,3-xylan. a The amount of reducing sugar released was measured by the Somogyi–Nelson method; 0.5% β-1,3-xylan in 50 mM sodium phosphate buffer (pH 7.5) were placed in Eppendorf tubes in the presence of 2.5 U ml−1 rTxyA and 0.15 U ml−1 rXloA either alone or in combination (total volume 0.3 ml). The reaction was assayed at 37°C. Each value represents the averages of at least duplicate measurements. Symbols: circles co-action of rTxyA and rXloA, squares action of rTxyA alone, triangles action of rXloA alone. b TLC analysis of hydrolysis products released from β-1,3-xylan. The reaction was carried out at 37°C for 12 h. Lane M standard oligosaccharides, lane 1 hydrolysis products by the action of rXloA, lane 2 rTxyA, lane 3 rTxyA and rXloA. TX2 β-1,3-xylobiose, TX3 β-1,3-xylotriose, TX4 β-1,3-xylotetraose, respectively
Isomerization of d-Xylose to d-Xylulose
d-Xylose produced from β-1,3-xylan was converted to d-xylulose by using rXylA because S. cerevisiae is unable to ferment d-xylose but can ferment d-xylulose. Figure 6 shows the time course of d-xylulose production from d-xylose by the purified rXylA at 40°C and pH 7.5. After the enzyme reaction was stopped by boiling the tubes, sugar composition was analyzed by HPLC. Although XIs are the enzymes that can isomerize d-xylose to d-xylulose, the equilibrium ratio of d-xylose/d-xylulose is typically 80:20 (Chandrakant and Bisaria 2000). In concordance with this, the conversion rate of d-xylose to d-xylulose by rXylA was found to be approximately 15% after 2 h of incubation. On the other hand, the conversion rate was increased up to around 40% when 4 mM sodium tetraborate was added to the reaction mixture. We investigated the effect of borate ion concentration against the conversion rate of xylose to xylulose. When the xylose isomerase activity of the XylA was measured on the reaction mixtures containing several concentration of sodium tetraborate (0, 1, 2, 4, 6, and 8 mM), the activity increased lineally to 4 mM concentration and arrived at plateau more than 4 mM. Therefore, 4 mM sodium tetraborate was added to the reaction mixture.

The time course of d-xylulose production from d-xylose by rXylA. The reaction mixture containing 50 mM sodium phosphate buffer (pH 7.5), 1.5% d-xylose, 10 mM MgSO4, 4 mM sodium tetraborate, and 0.25 U rXylA in Eppendorf tubes (total volume 1 ml) were incubated at 40°C. Symbols: triangles isomerization in the absence of sodium tetraborate, circles isomerization in the presence of 4 mM sodium tetraborate. Each value represents the mean of duplicate measurements
Discussion
The killer alga, mutant C. taxifolia, has spread uncontrollably in the Mediterranean Sea since 1984 and has destroyed local ecosystems in these waters. In this study, we tried to develop a method to produce ethanol from β-1,3-xylan, which is a major structural polysaccharide component of the C. taxifolia cell wall. To date, we have cloned and characterized the genes encoding TxyA (Araki et al. 2000) and XloA (Umemoto et al. 2008) from the marine bacterium Vibrio sp. strain XY-214. These two enzymes are required for the saccharification of β-1,3-xylan to d-xylose. Because yeast S. cerevisiae cannot ferment d-xylose but can utilize d-xylulose, conversion of d-xylose to d-xylulose by using a d-xylose isomerase is required after the enzymatic saccharification of β-1,3-xylan. In our previous study of the cloning of the xloA gene from the strain XY-214, we have obtained a truncated gene (xylA) encoding a XylA in the upstream region of the xloA gene. In the present study, therefore, we first cloned the complete xylA gene from the strain XY-214 and characterized the gene product. Second, we produced d-xylulose from β-1,3-xylan by using three kinds of enzymes (TxyA, XloA, and XylA).
Cloning of the xylA gene and the sequence analysis revealed a gene cluster involved in the β-1,3-xylan degradation and d-xylose metabolism in the genomic DNA of Vibrio sp. strain XY-214 (Fig. 1). The gene cluster is 11,101-bp long and contains eight genes organized in the order xylR, xylT, xylA, xloA, gatA, alrA, txyA, and xbpA. To our knowledge, this is the first identification of a β-1,3-xylan utilization gene cluster, although there have been many reports regarding the gene cluster involved in the utilization of β-1,4-xylan (Shulami et al. 1999; Erlandson et al. 2001; Chow et al. 2007). In most cases, the d-xylose isomerase gene forms an operon with a gene encoding d-xylulose kinase (xylB), which converts d-xylulose to d-xylulose-5-phosphate (Lawlis et al. 1984; Lokman et al. 1991; Sizemore et al. 1991; Wong et al. 1991; Feldmann et al. 1992). However, we could not detect the xylB gene in the upstream and downstream of the xylA gene. This indicates that the xylA and xylB genes are transcribed separately in case of the strain XY-214. On the other hand, the gatA and alrA genes probably form an operon because they were separated by only 32-bp nucleotides and no transcription terminator was detected between these two genes. The deduced amino acid sequence of the gatA gene showed similarities to a GAT from E. coli (accession no. CP000243; 43% identity). GAT is encoded by the lacA gene of the lac operon of E. coli. The lacA gene is cotranscribed with the lacZ and lacY genes which encode β-galactosidase and lactose permease, respectively. Although the roles of the latter two enzymes in lactose metabolism have been clearly characterized, the cellular function of GAT remains unclear (Lewendon et al. 1995). The properties of the alrA gene product have been investigated previously (Umemoto and Araki 2008). It has been revealed that an AlrA encoded by the alrA gene belongs to the aldo-keto reductase superfamily although the cellular functions of AlrA also remain unclear. Therefore, further biochemical studies are required to understand the roles of GAT and AlrA in the d-xylose metabolism. In order to understand the metabolic mechanism of β-1,3-xylan in Vibrio sp. strain XY-214, functional analysis of transcriptional regulation of the gene cluster will be required.
The xylA gene of Vibrio sp. strain XY-214 has been cloned and expressed in E. coli. Sequence analysis revealed that XylA is classified into the group II XIs, which differ from the group I XIs by the presence of approximately 50 additional residues at the N terminus. Although group II XIs share only 20–30% amino acid sequence identity with group I XIs, the active-site residues in group I and group II enzymes are highly conserved (Park and Batt 2004). Sequence alignment with other XIs revealed that all of the active-site residues and the residues having a role in the subunit interaction were conserved in XylA (Fig. 2). Similar to most of XIs reported so far, XylA of the strain XY-214 has been found to be a homotetramer with a molecular mass of 49,264 Da per subunit, whereas some XIs have been found to be dimeric (Batt et al. 1990; Meng et al. 1993). To date, the crystal structures of several group I XIs have been extensively characterized (Farber et al. 1987; Rey et al. 1988; Carrell et al. 1989; Henrik et al. 1989; Dauter et al. 1990; Whitlow et al. 1991; Rasmussen et al. 1994), and it has been revealed that each monomer of XIs consists of an (α/β)8 barrel having an active site and a C-terminal loop lacking β-strands (Park and Batt 2004). The XylA was activated strongly by magnesium ion. Allen et al. describe that magnesium ion is essential for isomerization but not essential for ring opening of d-xylose isomerase structure (Allen et al. 1994). As do other XIs, XylA has a lower K m for d-xylose (7.93 mM) than for d-glucose (187 mM). Although XylA shares common properties to most XIs from mesophilic bacteria, this is the first report on the characterization of XIs from Vibrio spp.
For the production of d-xylulose from β-1,3-xylan, the saccharification of β-1,3-xylan and isomerization of d-xylose to d-xylulose are required. In this study, each process was performed separately. Saccharification of β-1,3-xylan to d-xylose was carried out by using rTxyA and rXloA at 37°C and pH 7.5. As shown in Fig. 5b, d-xylose was produced as the major hydrolysis product by the co-action of rTxyA and rXloA, whereas the action of rTxyA alone predominantly produced β-1,3-xylobiose, with d-xylose and β-1,3-xylotriose as minor products. This indicated that rTxyA and rXloA are useful tools for the saccharification of β-1,3-xylan. d-Xylose prepared from β-1,3-xylan was then converted to d-xylulose by using rXylA at 40°C and pH 7.5. The conversion rate of d-xylose to d-xylulose reached approximately 15% after 2 h of incubation in the absence of sodium tetraborate. On the other hand, the conversion rate increased up to approximately 40% in the presence of 4 mM sodium tetraborate (Fig. 6). On the other hand, the conversion rate increased up to approximately 40% in the presence of 4 mM sodium tetraborate because borate ion is combined with xylulose and forms a complex. As a result, free xylulose decreases in the reaction mixture and the conversion efficiency of xylose to xylulose rises (Fig. 6).
In summary, we successfully produced d-xylulose from β-1,3-xylan using three kinds of bacterial enzymes by two steps processing. We are going to develop technology to produce xylulose from β-1,3-xylan directly by one-step method with these enzymes. We have also confirmed that d-xylulose produced from β-1,3-xylan can be fermented to ethanol by yeast S. cerevisiae (data not shown). While further studies are required to improve the conversion rate of β-1,3-xylan to d-xylulose, the results of the present study will serve as the basis for the ethanol production from the killer alga C. taxifolia.
References
Allen KN, Lvie A, Glasfeld A, Tanada TN, Gerrity DP, Carlson SC, Farber GK, Petsko GA, Ringe D (1994) Role of the divalent metal ion in sugar binding, ring opening, and isomerization by D-xylose isomerase: replacement of a catalytic metal by an amino acid. Biochemistry 33:1488–1494
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Aoki T, Araki T, Kitamikado M (1988) Purification and characterization of an endo-β-1,3-xylanase from Vibrio sp. Nippon Suisan Gakkaishi 54:277–281
Araki T, Inoue N, Morishita T (1998) Purification and characterization of β-1,3-xylanase from a marine bacterium, Alcaligenes sp. XY-234. J Gen Appl Microbiol 44:269–274
Araki T, Tani S, Maeda K, Hashikawa S, Nakagawa H, Morishita T (1999) Purification and characterization of β-1,3-xylanase from a marine bacterium, Vibrio sp. XY-214. Biosci Biotechnol Biochem 63:2017–2019
Araki T, Hashikawa S, Morishita T (2000) Cloning, sequencing, and expression in Escherichia coli of the new gene encoding β-1,3-xylanase from a marine bacterium, Vibrio sp. strain XY-214. Appl Environ Microbiol 66:1741–1743
Bailey RW, Bourne EJ (1960) Colour reagents given by sugars and diphenylamineaniline spray reagents on paper chromatograms. J Chromatogr 4:206–213
Batt CA, Jamieson AC, Vandeyar MA (1990) Identification of essential histidine residues in the active site of Escherichia coli xylose (glucose) isomerase. Proc Natl Acad Sci USA 87:618–622
Boraston AB, Bolam DN, Gilbert HJ, Davies GJ (2004) Carbohydrate-binding modules: fine tuning polysaccharide recognition. Biochem J 382:769–781
Carrell HL, Glusker JP, Burger V, Manfre F, Tritsch D, Biellmann JF (1989) X-ray analysis of D-xylose isomerase at 1.9 A: native enzyme in complex with substrate and with a mechanism-designed inactivator. Proc Natl Acad Sci USA 86:4440–4444
Chandrakant P, Bisaria VS (2000) Simultaneous bioconversion of glucose and xylose to ethanol by Saccharomyces cerevisiae in the presence of xylose isomerase. Appl Microbiol Biotechnol 53:301–309
Chen WP, Matsuo M, Yasui T (1986) Purification and some properties of β-1,3-xylanase from Aspergillus terreus A-07. Agric Biol Chem 50:1183–1194
Chow V, Nong G, Preston JF (2007) Structure, function, and regulation of the aldouronate utilization gene cluster from Paenibacillus sp. strain JDR-2. J Bacteriol 189:8863–8870
Dalton R (2000) Researchers criticize response to killer algae. Nature 406:447
Dauter Z, Terry H, Witzel H, Wilson KS (1990) Refinement of glucose isomerase from Streptomyces albus at 1.65 Å with data from an imaging plate. Acta Crystallogr B 46:833–841
Dische Z, Borenfreund EA (1951) A new spectrophotometric method for the detection and determination of keto sugars and trioses. J Biol Chem 192:583–587
Erlandson KA, Soazig Delamarre C, Batt CA (2001) Genetic evidence for a defective xylan degradation pathway in Lactococcus lactis. Appl Environ Microbiol 67:1445–1452
Farber GK, Petsko GA, Ringe D (1987) The 3 Å crystal structure of xylose isomerase from Streptomyces olivochromogenes. Protein Eng 1:459–466
Feldmann SD, Sahm H, Sprenger GA (1992) Cloning and expression of the genes for xylose isomerase and xylulokinase from Klebsiella pneumoniae 1033 in Escherichia coli K12. Mol Gen Genet 234:201–210
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788
Gong CS, Chen LF, Flickinger MC, Chiang LC, Tsao GT (1981) Production of ethanol from D-xylose by using D-xylose isomerase and yeasts. Appl Environ Microbiol 41:430–436
Henrik K, Collyer CA, Blow DM (1989) Structures of D-xylose isomerase from Arthrobacter strain B3728 containing the inhibitors xylitol and D-sorbitol at 2.5 Å and 2.3 Å resolution, respectively. J Mol Biol 208:129–147
Hess JM, Tchernajenko V, Vielle C, Zeikus JG, Kelly RM (1998) Thermotoga neapolitana homotetrameric xylose isomerase is expressed as a catalytically active and thermostable dimer in Escherichia coli. Appl Environ Microbiol 64:2357–2360
Hsiao HY, Chiang L, Chen L, Tsao GT (1982) Effect of borate on isomerisation and yeast fermentation of high xylulose solution and acid hydrolysate of hemicellulose. Enzyme Microb Technol 4:25–31
Iriki Y, Suzuki T, Nisizawa K, Miwa T (1960) Xylan of siphonaceous green algae. Nature 187:82–83
Jousson O, Pawlowski J, Zaninetti L, Meinesz A, Boudouresque CF (1998) Molecular evidence for the aquarium origin of the green alga Caulerpa taxifolia introduced to the Mediterranean Sea. Mar Ecol Prog Ser 172:275–280
Kaiser L (2000) California algae may be feared European species. Science 289:222–223
Kiyohara M, Sakaguchi K, Yamaguchi K, Araki T, Nakamura T, Ito M (2005) Molecular cloning and characterization of a novel β-1,3-xylanase possessing two putative carbohydrate-binding modules from a marine bacterium Vibrio sp. strain AX-4. Biochem J 388:949–957
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lawlis VB, Dennis MS, Chen EY, Smith DH, Henner DJ (1984) Cloning and sequencing of the xylose isomerase and xylulokinase genes of Escherichia coli. Appl Environ Microbiol 47:15–21
Lewendon A, Ellis J, Shaw WV (1995) Structural and mechanistic studies of galactoside acetyltransferase, the Escherichia coli LacA gene product. J Biol Chem 270:26326–26331
Lin Y, Tanaka S (2006) Ethanol fermentation from biomass resources: current state and prospects. Appl Microbiol Biotechnol 69:627–642
Lokman BC, Santen PV, Verdoes JC, Kruse J, Leer RJ, Posno M, Pouwels PH (1991) Organization and characterization of the three genes involved in D-xylose catabolism in Lactobacillus pentosus. Mol Gen Genet 230:161–169
Lowry OH, Rosebrouigh NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
McDowell RH (1967) Chemistry and enzymology of marine algal polysaccharides. Academic, London, pp 88–96, 134–137
Meinesz A, Hesse B (1991) Introduction et invasion de l’algue tropicale Caulerpa taxifolia en Méditerranée nord-occidentale. Oceanol Acta 14:415–426
Meinesz A, Belsher T, Thibaut T, Antolić B, Ben Mustapha K, Boudouresque CF, Chiaverini D, Cinelli F, Cottalorda JM, Djellouli A, El Abed A, Orestano C, Grau AM, Ivesa L, Jaklin A, Langar H, Massuti-Pascual E, Peirano A, Tunesi L, de Vaugelas J, Zavodnik N, Zúljević A (2001) The introduced green alga Caulerpa taxifolia continues to spread in the Mediterranean. Biol Invasions 3:201–210
Meng M, Lee C, Bagdasarian M, Ziekus JG (1991) Switching substrate preference of thermophilic xylose isomerase from D-xylose to D-glucose by redesigning the substrate binding pocket. Proc Natl Acad Sci USA 88:4015–4019
Meng M, Bagdasarian M, Zeikus JG (1993) Thermal stabilization of xylose isomerase from Thermoanaerobacterium thermosulfurigenes. Bio/Technology 11:1157–1161
Meusnier I, Olsen JL, Stam WT, Destombe C, Valero M (2001) Phylogenetic analyses of Caulerpa taxifolia (Chlorophyta) and of its associated bacterial microflora provide clues to the origin of the Mediterranean introduction. Mol Ecol 10:931–946
Okazaki F, Tamaru Y, Hashikawa S, Li YT, Araki T (2002) Novel carbohydrate-binding module of β-1,3-xylanase from a marine bacterium, Alcaligenes sp. strain XY-234. J Bacteriol 184:2399–2403
Olsen JL, Valero M, Meusnier I, Boele-Bos S, Stam WT (1998) Mediterranean Caulerpa taxifolia and C. mexicana (Chlorophyta) are not conspecific. J Phycol 34:850–856
Park JH, Batt CA (2004) Restoration of a defective Lactococcus lactis xylose isomerase. Appl Environ Microbiol 70:4318–4325
Rangarajan M, Hartley BS (1992) Mechanism of D-fructose isomerization by Arthrobacter D-xylose isomerase. Biochem J 283:223–233
Rasmussen H, Cour TL, Nyborg J, Schulein M (1994) Structure determination of glucose isomerase from Streptomyces murinus at 2.6 Å resolution. Acta Crystallogr Sect D 50:124–131
Rey F, Jenkins J, Janin J, Lasters I, Alard P, Claessens M, Matthyssens G, Wodak S (1988) Structural analysis of the 2.8 Å model of xylose isomerase from Actinoplanes missouriensis. Proteins Struct Funct Genet 4:165–172
Rosenberg M, Court D (1979) Regulatory sequences involved in the promotion and termination of RNA transcription. Annu Rev Genet 13:319–353
Schaffelke B, Murphy N, Uthicke S (2002) Using genetic techniques to the sources of the invasive alga Caulerpa taxifolia in three new locations in Australia. Mar Pollut Bull 44:204–210
Shulami S, Gat O, Sonenshein AL, Shoham Y (1999) The glucuronic acid-utilization gene cluster from Bacillus stearothermophilus T-6. J Bacteriol 181:3695–3704
Sizemore C, Buchnere E, Rygus T, Witke C, Goetz F, Hillen W (1991) Organization, promoter analysis and transcriptional regulation of the Staphylococcus xylosus xylose utilization operon. Mol Gen Genet 227:377–384
Somogyi M (1952) Notes on sugar determination. J Biol Chem 195:19–23
Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamura H, Ohmichi T, Yoneyama M, Sasaki M (1995) Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34:11211–11216
Umemoto Y, Araki T (2008) Expression in Escherichia coli and characterization of an aldehyde reductase from a marine bacterium, Vibrio sp. strain XY-214. Bull graduate school of bioresources. Mie Univ 35:47–53
Umemoto Y, Onishi R, Araki T (2008) Cloning of a novel gene encoding β-1,3-xylosidase from a marine bacterium, Vibrio sp. strain XY-214, and characterization of the gene product. Appl Environ Microbiol 74:305–308
Whitlow M, Howard AJ, Finzel BC, Poulos TL, Winborne E, Gilliland GL (1991) A metal-mediated hydride shift mechanism for xylose isomerase based on the 1.6 Å Streptomyces rubiginosus structures with xylitol and D-xylose. Proteins Struct Funct Genet 9:153–173
Wong HC, Ting Y, Lin HC, Reichert F, Myambo K, Watt KWK, Toy PL, Drummond RJ (1991) Genetic organization and regulation of the xylose degradation genes in Streptomyces rubiginosus. J Bacteriol 173:6849–6858
Yamaura I, Matumoto T, Funatsu M, Murai E (1990) Purification and some properties of endo-β-1,3-xylanase from Pseudomonas sp. PT-5. Agric Biol Chem 54:921–926
Acknowledgments
This work was supported by the JSPS Research Fellowships for Young Scientists (No. 21-5436 to Y.U.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Umemoto, Y., Shibata, T. & Araki, T. d-Xylose Isomerase from a Marine Bacterium, Vibrio sp. Strain XY-214, and d-Xylulose Production from β-1,3-Xylan. Mar Biotechnol 14, 10–20 (2012). https://doi.org/10.1007/s10126-011-9380-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-011-9380-9