
Abstract
The genetic relationships among northern South China Sea populations of the six bar wrasse (Thallasoma hardwicki) were investigated. Fish collected from the Solomon Islands were used for geographic comparison. In 1998 and 1999, a total of 100 fish were sampled from 6 localities of the northern South China Sea and 3 localities of the Solomon Islands. Genetic variations in DNA sequences were examined from the first hypervariable region (HVR-1) of the mitochondrial control region, as amplified by polymerase chain reaction. High levels of haplotypic diversity (h = 0.944 ± 0.0016, π = 0.0224 ± 0.01171) in the HVR-1 region of the mitochondrial control region of T. hardwicki were detected. This yielded 94 haplotypes that exhibited a minimum spanning tree with a starburst structure, suggestive of a very recent origin for most haplotypes. Neutrality tests indicated that the pattern of genetic variability in T. hardwicki is consistent either with genetic hitchhiking by an advantageous mutation or with population expansion. Partitioning populations into coherent geographic groups divided the northern South China Sea samples (ΦCT = 0.0313, P < 0.001) into 3 major groups: a north-central group composed of northwestern Taiwan and northern Vietnam; a southwestern group containing southern Vietnam; and a southern group including the central Philippines. These results are in concordance with mesoscale boundaries proposed by allozyme markers, thus highlighting the importance of identifying transboundary units for the conservation and management of fisheries in the South China Sea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The literature is replete with examples of marine fish species with high levels of gene flow that exhibit no genetic structure over geographic scales spanning hundreds to thousands of kilometers (Palumbi, 1992, 1994; Benzie, 1998, 1999; Waters et al., 2000; Riginos and Victor, 2001; Planes and Fauvelot, 2002). Genetic homogeneity across such large geographic scales is usually explained by the connections created by sea surface currents. In contrast, some studies have documented that reef fishes with high dispersal capacities exhibit high genetic divergence among populations in the absence of distinct barriers to dispersal (Bell et al., 1982; Planes et al., 1994; McMillian and Palumbi, 1995; Shulman and Berminghan, 1995; Waters et al., 2000; Riginos and Victor, 2001; Fauvelot and Planes, 2002; Planes and Fauvelot, 2002; Taylor and Hellberg, 2003). Palumbi (1996b) reviewed 4 possible explanations for divergence among marine populations: behavior mechanisms (e.g., vertical migration of larval or adult fishes; Planes et al., 1994); selection against immigrants or selection for balanced polymorphisms; complex oceanographic circulation patterns; and historic barriers to gene flow.
The South China Sea, extending 2800 km north from the Equator to the Tropic of Cancer and averaging 1000 km in width, harbors the highest diversity of marine species anywhere on Earth (reviewed in McManus, 1992, 1997). The South China Sea is subject to monsoonal wind patterns that shift semiannually from origins in the northeast to origins in the southwest (Figure 1, reviewed in McManus, 1992). This results in a complex system of surface currents, many of which completely reverse on an annual or semiannual basis (Figure 1). Whether these complex surface currents determine the genetic structure of marine organisms within the South China Sea is still poorly understood. Some have suggested that historic barriers, such as sea level changes during the Pleistocene, may have played important roles in creating isolated populations by cutting off local sea basins from the Southeast Asian region (McManus, 1985; Potts, 1985), and the resulting structure persists to this day. It is important to understand the extent to which present conditions or historical events have determined the current genetic structure within a species, because genetic data are interpreted as indirect estimates of dispersal in marine organisms. Genetic structures and the time frame for the mechanism that brought them about have implications for the conservation and sustainable management of these fisheries.

Surface currents of the South China Sea in winter (a) and summer (b).
Evidence from a large-scale allozyme study of the damselfish Dascyllus trimaculatus, using 12 markers from 16 sites in 6 countries (Indonesia, Malaysia, the Philippines, Taiwan, Vietnam, and the Solomon Islands), suggested the occurrence of 4 major regional management units in the South China Sea and adjacent areas: (1) a West Pacific group to the east of the Philippines and southeast of Taiwan; (2) a north-central group encompassing northwestern Taiwan, northern Vietnam, and the northwestern Philippines; (3) a southwestern group comprising southern Vietnam and the eastern coast of mainland Malaysia; and (4) a southern group including the southern and central Philippines, eastern Malaysia, and central Indonesia (Ablan et al., 2002). The extent to which this pattern occurs in other reef fish species, particularly a species with highly contracting reproductive strategies and much longer larval durations, will provide more insights into the validity of the suggested regional management boundaries for reef-associated fisheries resources.
To expand our understanding of the genetic structure of reef fish populations in the South China Sea, we analyzed the DNA sequences from the first hypervariable region (HVR-1) of the mitochondrial control region in six bar wrasse (Thallasoma hardwicki) collected from 6 populations of 3 countries in the northern South China Sea—the Philippines, Taiwan, and Vietnam. In addition, fish from 3 populations of the Solomon Islands were collected for geographic comparison. Thallasoma hardwicki is a widely distributed and frequently observed species of reef fish in the South China Sea (Froese and Pauly, 2002). Any finding of concordant or discordant genetic structure of T. hardwicki in the 4 proposed management units within the region would be of considerable practical significance for the conservation and management of fisheries in the South China Sea.
MATERIALS AND METHODS
Fish Samples
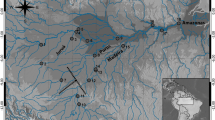
Fish were collected through an international collaborative project among 6 countries around the South China Sea and the Solomon Islands, coordinated by the World Fish Center in 1997. In total, 100 Thallasoma hardwicki individuals were collected from 6 populations in the northern South China Sea and from 3 populations of the Solomon Islands (Figure 2, Table 1). Thallasoma lunare, T. quinquvevittatum, and T. purpureum were used as outgroups for phylogenetic analysis. Fish were frozen whole in liquid nitrogen and shipped to the headquarters of the World Fish Center for allozyme electrophoretic analysis (M.C.A., Ablan et al., unpublished data) Subsequently, a piece of muscle tissue was removed, preserved in 95% ethanol, and shipped to the laboratory of the senior author at the Institute of Zoology, Academia Sinica, Taiwan, for DNA analysis.

Locations of the 6 Thallasoma hardwicki populations across the northern South China Sea and the 3 populations in the Solomon Islands.
DNA Extraction, Amplification, and Sequencing
DNA extraction was modified from methods described in Chen and Yu (2000) and Chen et al. (2000). A 400-bp portion of HVR-1 in the mitochondrial control region was amplified by the polymerase chain reaction (PCR) using a pair of specific primers, THDIoop-F, 5′-AGC ACC GGT CTT GTA AAC CGG CAG-3′, and THDloop-R420, 5′-GTC CTG GTT GGT CGG TTC TTA CTG CG-3′, designed from the complete DNA sequence of the mitochondrial control region of T. hardwicki (C.A. Chen, unpublished data). PCR was performed in a PC-9606 thermal sequencer (Corbett Research, Australia) using the following thermal cycle: 1 cycle at 95°C (4 minutes); 4 cycles at 94°C (30 seconds), 50°C (1 minute), and 72°C (2 minutes), followed by 35 cycles at 94°C (30 seconds), 55°C (1 minute), and 72°C (2 minutes). The amplification reaction used 50 to 200 ng of the template and Taq polymerase (BRL) in a 50-μl-volume reaction using the buffer supplied with the enzyme and under the conditions recommended by the manufacturer. Each 50-μl reaction volume contained 50 mM KCl, 10 mM Tris (pH 9.0), 1 .5 mM MgCl2, 0.2 mM dNTPs, 0.33 unit of each primer, 0.033 units of Taq polymerase, and 100 ng of genomic DNA. The PCR products were electrophoresed in a 1% agarose (FMC Bioproduct) gel in 1× TAE buffer to check the yield. The amplified DNA was extracted using a DNA/RNA gel extraction column (Viogene, Taipei) under conditions recommended by the manufacturer. The nucleotide sequences were determined for both ends of the PCR products using an ABI 377 Genetic Analyzer. The sequences obtained in this study have been submitted to GenBank under accession numbers AY185923 to AY186029.
Data Analyses
All Thallasoma hardwicki sequences were initially aligned using CLUSTAL W 1.7 (Thompson et al., 1994), followed by manual editing using SeqApp 1.9 (Gilbert, 1994). All data analyses were implemented using Arlequin 2.0 for Macintosh platform (Schneider et al., 2000). The unique haplotypes and transitions and transversions were counted. The haplotypic diversity, h, nucleotide diversity, π (Nei, 1987), and their standard errors were calculated. We also constructed haplotype networks for T. hardwicki. Matrices of the absolute number of substitution differences were produced. The minimum spanning tree (MST; Excoffier, 1993) of haplotypes, drawn by hand, was based on the output. The historical demography of T. hardwicki was examined using mismatch distributions based on the observed number of differences among all pairs of haplotypes. Simulation studies have shown that distribution is multimodal in samples drawn from populations at demographic equilibrium, a pattern reflecting stochastic lineage loss. In contrast, distribution appears unimodal in populations that have passed through recent demographic expansions within the time horizon of the marker (Slatkin and Hudson, 1991; Roger and Harpending, 1992). We also used Tajima’s D, Fu’s F statistic (Fu and Li, 1993), and Fu’s Fs (Fu, 1996) to further examine the historical demography of T. hardwicki (Tajima, 1989a, 1989b). If populations have experienced different modes of selection, demographic fluctuation, or population subdivision, then deviations from neutral expectations for the frequency of polymorphisms are expected. Tajima’s D can be used to examine demography in that a negative value is expected under population expansion and a positive value is expected under population subdivision. Fu’s F statistic and Fs statistic can detect the effect of population growth, i.e., an excess of low-frequency mutations. Fu’s Fs statistic is more sensitive to genetic hitchhiking and population growth, while Fu’s F statistic is more sensitive to background selection (Fu, 1996, 1997). The estimated demographic parameters of each population were then used as input in a coalescent simulation program to generate DNA sequences (Schneider and Excoffier, 1999) for which new sets of parameters were computed. Approximate 95% confidence intervals were generated after 5000 simulations for the mismatch distribution and the number of polymorphic sites. Several parameters, including τ, θ0, and θ1, were calculated: τ is the age of expansion, while θ0 and θ1 are the population sizes before and after expansion, respectively. P(SSDobs) is the probability of observation by chance of a less than good fit between the observed distribution and the mismatch distribution for a demographic history of a population defined by the estimated parameters τ, θ0, and θ1 (Petit et al., 1999). The mismatch distribution and coalescent simulation were computed.
In order to reveal the population genetic structure of Thallasoma hardwicki in the northern South China Sea and its relationship to that in the Solomon Islands, we conducted two statistical analyses. First, total genetic variations of the northern South China Sea and the Solomon Islands populations were computed on the basis of the haplotype frequency distribution analysis (equivalent to a Fst analysis; Cockerham, 1973) corrected for interhaplotype sequence divergence using the Kimura 2-parameter model (Kimura, 1980). A Φ st value, analogous to the Cockerham Fst estimation of genetic variation, was calculated to estimate the genetic divergence overall and between paired populations. The statistical significance of the total and pairwise Φ st values was estimated by comparison of the observed distribution generated by 10,000 permutations, in which individuals are randomly redistributed into populations. The exact test of population differentiation (Raymond and Rousset, 1995) based on haplotype frequencies was also applied as implemented using Markov chain length, 20,000 steps. The significance threshold of pairwise comparisons (P < 0.05) was always adjusted by sequential Bonferroni correction (Rice, 1989).
Second, the extent of population subdivision between populations was examined using analysis of molecular variance (AMOVA; Excoffier et al., 1992). The standard variance and haplotypic correlation measures (Φ statistics) can be used to reveal the degree of population subdivision (Schneider et al., 2000). To correct for any effects of unequal nucleotide composition, multiple substitutions, and differences in substitution rates among sites in the control region that are characteristic of this region (see Perna and Kocher, 1995; Rosel and Block, 1996), interhaplotypic distances for AMOVA were estimated using the Tamura-Nei evolution model (Tamura and Nei, 1993). The shape parameter, α, of a γ distribution of mutation rates was estimated using the method of moments (Johnson and Kotz, 1975), as described by Petit et al. (1999). The significance of the resultant statistics and variance components was tested with 1 0,000 permutations.
RESULTS
Control Region Sequence Variation
After unreadable sites close to the primers at either end were removed, 332 bp of HVR-1 sequences were obtained for 100 Thallasoma hardwicki from 9 populations. In a total of 94 haplotypes with 91variable sites and 41 phylogenetically informative sites were identified for T. hardwicki (Table 1). The nucleotide composition was A+T-rich (A, 41.5%; T, 29.2%), and variations consisted predominantly of transition substitutions (Ti: Tv = 4.0). A γ value of 0.18 was calculated for T. hardwicki, suggesting strong heterogeneity of substitution rates among nucleotide sites in its HVR-1. The greatest number of absolute differences among haplotypes was 8. Total haplotypic diversity was high (0.94 ± 0.0016). Of the 9 populations, all except Zambales had a haplotypic diversity of 1; i.e., in most of populations, there was no identical haplotype shared by T. hardwicki within the population (Table 1).
Pattern of Historical Demography
Relationships between haplotypes are represented on a minimum spanning tree (MST), and the MST of T. hardwicki appeared star-like, with many individual haplotypes linked by a single or a few mutations (Figure 3). Among the 94 haplotypes, only 1 (c6/w3) was shared between the South China Sea and Solomon Island population. The MST showed no obvious haplotype clustering with respect to the South China Sea and the Solomon Islands. These characteristics of the T. hardwicki mitochondrial control region are consistent with results from other studies assessing fish mitochondrial-control-region variability (Lee et al., 1995; Dudgeon et al., 2000).

Thallasoma hardwicki haplotype network. Each circle represents a unique haplotype; small circles represent 1 individual and large ones represent 2 individuals possessing that haplotype. White circles represent the South China Sea haplotype; hatched circles, the Solomon Island haplotype; and dotted circle, the haplotype shared by both regions. Abbreviations of localities are given in Table 1.
The high haplotypic diversity (0.998 ± 0.0016, mean ± SE) relative to nucleotide diversity (0.0224 ± 0.01171) is indicative of a population bottleneck followed by rapid population growth and accumulation of mutations (Grant and Bowen, 1998; Avise, 2000). Thallasoma hardwicki has a mismatch distribution that fits well with a model of population expansion (Figure 4). This is supported by a significant negative value for Tajima’s D (Table 2) for all populations (−1.61784, P < 0.05). In addition, Fu’s Fs statistic, which is more sensitive to genetic hitchhiking and population growth, was significant in all populations. In contrast, Fu’s F statistic, which is more sensitive to background selection, was never significant (Table 2). We also noticed that Fs was negative in all populations but 2—the western population of the Solomon Islands and that from Zambales in the Philippines had positive values. Thallasoma hardwicki has a mismatch distribution that fits well with a model of population expansion (Figure 4). The estimation of θ0, and θ1, and the P values of the SSDobs statistic revealed that the patterns of polymorphism in 7 of 8 populations (except Zambales) were under a population expansion model (Table 3). For the Zambales population, significantly small P values of SSDobs indicated that the pattern of polymorphism observed in this population was under a stationary population model. For the 7 populations for which a model of population expansion was indicated by the SSDobs test, the observed number of polymorphic sites (s) was within the range of the 95% confidence interval generated by the simulations (Si), suggesting that the parameters estimated by the model accurately accounted for the polymorphism observed in these populations (Table 3).

Mismatched distribution of Thallasoma hardwicki pairwise sequence differences as computed by ARLEQUIN 2.0.
Patterns of Population Structure
The population structure was estimated from 8 populations; Lanyu was excluded from the following analyses because of its small sample size. Most of the pairwise Φ st values (Table 4) were low and statistically not significant (25 of 28). However, all the Φ st values observed were not significantly different after application of the sequential Bonferroni correction (P = 0.3174), suggesting a pattern of overall mitochondrial control region homogeneity of the South China Sea and the Solomon Island Thallasoma hardwicki. In addition, we examined the population structure using 3 sets of hierarchical AMOVAs. First, we divided the groups according to natural geographic boundaries: i.e., Penghu, the Philippines, and Vietnam. Second, the groups were assigned according to the mesoscale boundaries suggested by Ablan et al. (2002): i.e., the north-central group contains Penghu and Qui Nhon of Vietnam; the southwestern group contains Nha Trang and Ca Na Bay of Vietnam; and the southern group contains Zambales of the Philippines. Finally, we compared genetic structures between the populations in the northern South China Sea and those in the Solomon Islands (Table 5). The AMOVA, incorporating both sequence divergence and haplotype frequencies among populations, detected significant structuring among populations with in the northern South China Sea using mesoscale boundaries as grouping criterion (Φ CT = 0.0313, P < 0.001). Population subdivision was not significant using natural geographic boundaries as the criterion (Φ CT = 0.01795, P > 0.01), or was it significant between the Solomon Islands and the South China Sea (Φ CT = 0.01136, P > 0.01). Within-population variations were all significant in the 3 comparisons, with the greatest variation in genetic structure (97.66%) found in the comparison between the South China Sea and the Solomon Islands. Negative values were obtained for some comparisons. Negative values indicate great differences between 2 random individuals from the same population, rather than between 2 random individuals from different populations (Arnason and Palsson, 1996).
DISCUSSION
Variability of the HVR-1 Region in Thallasoma hardwicki
Thallasoma hardwicki showed high levels of haplotypic diversity but relatively low sequence divergence in the HVR-1 region of the mitochondrial control region. Haplotype and nucleotide diversity estimates were similar to values reported for 2 scarid fishes, Scarus frenatus and Chlorurus sodidus (Dudgeon et al., 2000), and for several species of Caribbean reef fishes from mtDNA restriction fragment length polymorphism (RFLP) data (Shulman and Bermingham, 1995). Several scenarios have been proposed to explain the maintenance of high haplotypic diversity within populations, including large population sizes, environmental heterogeneity, and life-history traits that favor rapid population increase (Nei, 1987). In marine fishes, large population sizes (in excess of hundreds of millions of individuals) were thought to be responsible for extraordinarily high levels of genetic diversity (reviewed in Avise, 1998). Thallasoma hardwicki is a widely distributed and frequently observed species of reef fish in the South China Sea (Froese and Pauly, 2002), indicating that large population sizes may account for the high levels of haplotypic diversity observed in this study. A similar pattern was observed in the Caribbean congeneric species T. bifasciatum, as revealed by RFLP analysis of mtDNA (Shulman and Bermingham, 1995). In addition, life-history traits, such as short generation times, small maximum size, and quick maturation, have shown a significant correlation with high heterozygosity in bony fishes (Mitton and Lewis, 1989). This correlation was also proposed for the comparison between S. frenatus and C. sodidus, in which the former has a maximum life span that is 2 times longer than the latter. Chlorurus sodidus has greater haplotypic and nucleotide diversities than S. frenatus (Dudgeon et al., 2000). However, little is known about these life-history traits for T. hardwicki, and further work is needed to examine this correlation.
Patterns of Historical Demography
Results from the MST analysis and shallow divergence among Thallasoma hardwicki haplotypes suggest that this species has experienced a series of increases and reductions in population size throughout its history. In particular, population bottlenecks and expansions can produce the starburst pattern of haplotype relatedness (Figure 3) seen in T. hardwicki(Slatkin and Hudson, 1991; Roger and Harpending, 1992). The unimodal distribution of pairwise distances between haplotypes of T. hardwicki do fit a model of population expansion. This finding is corroborated by significantly negative Tajima’s D and Fs statistic, and by the high level of haplotypic diversity and low level of nucleotide diversity (Grant and Bowen, 1998). Although both D and Fs are consistent with population expansion (demographic hypothesis), the other alternative hypothesis, genetic hitchhiking by an advantageous mutation (selective hypothesis), should also be noted (Maruyama and Birky, 1991; Slatkin and Hudson, 1991; Fu, 1997). Deciding between the selective hypothesis and the demographic hypothesis requires comparison of the behavior of several unlinked loci (Tajima, 1989b). Unfortunately, we did not conduct this examination in the present study. Nevertheless, a good fit of observed mismatch distributions to the model of sudden population growth (Figure 4) supports the neutrality test of T. hardwicki populations (with the exclusion of Zambales), favoring the demographic hypothesis. Although the statistical analysis supports T. hardwicki populations having experienced a sudden expansion, evaluation of our model reveals that it is unable to account for the diversity of the Zambales population (Table 4). This is probably due to either an incorrect mutational model (Bertorelle and Slatkin, 1995; Aris-Brosou and Excoffier, 1996) or a demographic history other than a simple sudden expansion, such as an admixture of events as in some human populations (Marjoram and Donnelly, 1994).
Population Structure
We conclude from demographic analyses that Thallasoma hardwicki has experienced demographic changes throughout its history, and that the changes have been recent enough to remove much of the nucleotide divergence among haplotypes. In order to understand patterns of genetic structure, population genetic analyses that focus on a more recent time frame are needed. The pairwise Φ st statistics and AMOVA allow us to test for large-scale patterns of genetic structure. Even though MST failed to detect historical fragmentation of T. hardwicki, a genetic division might still exist among populations within the northern South China Sea. Indeed, the pairwise Φ st statistics and AMOVA did detect highly significant structuring among 5 populations within the South China Sea, but it was insignificant between the South China Sea and 3 populations in the Solomon Islands.
Three implications can be drawn from the results of pairwise Φ st statistics and AMOVA. First, genetic structuring detected in T. hardwicki is not concordant with the scenario that genetic heterogeneity of reef fishes is correlated with pelagic larval dispersal ability (Doherty et al., 1994, 1995; Shulman and Bermingham, 1995; Dudgeon et al., 2000; Riginos and Victor, 2001). It has been proposed that species with shorter pelagic phases tend to show significant mtDNA differences between populations, while species with longer pelagic durations do not. In the confamilial comparison of 2 Caribbean labrid fishes, Halichoeres bivittatus and Thallasoma bifasciatum, the former has a shorter pelagic duration (24 days; see Victor, 1986) and shows significant population structure by AMOVA, but the latter, with a longer larval life (49 days), does not (Shulman and Bermingham, 1995). The planktonic larval stage of T. hardwicki was estimated to be 47 days (Victor, 1986), which is similar to that of T. bifasciatum; therefore, it is expected that metapopulations would be maintained in a state of panmixia for T. hardwicki. However, this was not the case observed in the present study. Indeed, Shulman and Bermingham (1995) concluded that neither egg type nor length of larval duration is a simple predictor of geographic structure in reef populations after finding contrast results in 3 Caribbean fish species. Recently, results of a partial mitochondrial cytochrome b sequence suggested that populations of a Caribbean cleaner goby, Elacatinus evelynae, can remain demographically closed for thousands of generations despite extend larval duration (Taylor and Hellberg, 2003).
Second, AMOVA using allozyme markers (Ablan et al., 1999, 2002) and using 5′-end DNA sequence of the mitochondrial control region indicated a concordant genetic structure of T. hardwicki within the South China Sea. A 4-province division was proposed for the allozyme survey of the damselfish Dascycllus trimaculatus in this region, and a similar pattern was observed in T. hardwicki using allozyme markers, although sampling was not as extensive as for the damselfish (Ablan et al., 1999). According to Ablan’s model, AMOVA of T. hardwicki’s mitochondrial control region shows the highest percentage of total variance among regions (3.13%), and a more statistically significant Φ CT than those derived from natural geographic boundaries. This result supports the hypothesis that 4 subprovinces exist in the South China Sea, and the patterns observed are highly correlated with the general flow of sea surface currents (Figure 1) in the South China Sea (Ablan et al., 2002). Nevertheless, our interpretation should be considered preliminary because the number of sites and sample size per population surveyed in this study were limited. Future sampling from Malaysia, Indonesia, and other sites in the Philippines is needed to confirm this model.
Third, when the 3 populations from the Solomon Islands were included in the AMOVA, the percentage of total variance among regions dropped considerably, and Φ CT became insignificant. This result implies that gene flow between the northern South China Sea and Solomon Islands for T. hardwicki is high, and in concordance with major trends in several marine invertebrates (e.g., giant clams, Tridacna spp.) that have an axis trending northwest to southeast through the West Pacific (Benzie and Williams, 1995, 1997; reviewed in Benzie, 1998, 1999). However, this pattern is inconsistent with the present-day major sea surface currents between the Solomon Islands and the South China Sea. Therefore, larval dispersal by ocean currents alone cannot totally account for the high gene flow of T. hardwicki between the northern South China Sea and the Solomon Islands.
In contrast, currents at a depth of 100 m are more parallel to the island chains, and it has been proposed that the sea surface currents at the time of low sea levels may well have flowed from the northwest to the southwest (Benzie and Williams, 1995, 1997). This hypothesis is supported by the MST with a centralized haplotype, m5/g7, in the Solomon Islands, which may represent an ancestral haplotype to those in the South China Sea. In addition, only 1 haplotype, c6/w3, was shared by both the South China Sea and the Solomon Islands, indicating that existing gene flow between them might be limited. We concluded that patterns of spatial differentiation in giant clams, as ghosts of the dispersal past, are comparable to the pattern seen in T. hardwicki, and that these have not been changed by the system of currents since at least the early Holocene (Benzie and Williams, 1995, 1997; reviewed in Benzie, 1998, 1999).
Implications for Fisheries Management in the South China Sea
Marine ecosystems of the South China Sea are among the most heavily affected by human fishing in the world (Pauly et al., 1996; Silvestre and Pauly, 1997). Conservation of marine organisms within the South China Sea is a challenging enterprise. It covers a broad latitudinal range that makes the direct observation, study, and collection of marine species difficult (McManus, 1992, 1997). Overfishing on reefs limits the study of commercial fish to a few species that are common to all areas in sufficient numbers. Furthermore, this region is politically sensitive. A widely distributed noncommercial and reef-associated species, such as T. hardwicki, provides an opportunity to elucidate the connectivity of fish populations within the South China Sea.
Demographic expansion provides the historical mechanism for the formation and consequence of population structuring in T. hardwicki. It would be interesting to investigate the extent to which these models apply to other reef species in the region and to compare the results between exploited and unexploited species. That would allow us to validate the neutrality models under the unique situation of extinction-level exploitation rates, complex multigear and multispecies fisheries, similarity in target species over a wide range, the open access nature of fisheries, and the rapid coastal development and climate changes that may cause large-scale alternations in biophysical cycles.
In addition, the mitochondrial DNA sequence analyses adopted in the present study do provide a preliminary and feasible approach to determining population interdependencies around the South China Sea. Although the interpretation might be limited by the high mutation rate of the mitochondrial HVR-1, and sampling for this species was not as widespread as that for Dascyllus trimaculatus in Ablan et al. (2002), the results for the populations in this study are consistent with the proposed mesoscale boundary units suggested for the management of reef fisheries in the region, despite the fact that they represent the extreme range of reproductive strategies and pelagic larval duration of reef fish species. Dascyllus trimaculatus adults lay eggs in nests, and their larvae remain in the water column for 7 to 14 days. Thallasoma hardwicki adults are broadcast spawners, and the larvae settle after as many as 49 days. The results suggest that mesoscale management units proposed are sufficiently robust to apply to a wide range of reef fish species on a regional scale, and that population structures on a finer scale are likely to define units within these 4 regional management units. Investigations with a wider number of sites for T. hardwicki and a larger number of species, particularly for commercially important species, are required to test these hypotheses and their implications for the management of reef-associated fisheries. Sampling of T. hardwicki from other areas of the South China Sea and extension to the examination of concordant and discordant patterns from other taxa are therefore needed to provide complete information for the conservation and management of fisheries in the South China Sea.
References
Ablan, M.C.A., McManus, J.W., Chen, C.A., Shao, K.T., Bell, J., Cabanban, A.S., Tuan, V.S. and Arthana, I.W. (1999). Population interdependences in the South China Sea ecosystem. Technical Report (submitted to the John D. and Catherine T. MacArthur Foundation). Penang, Malaysia: ICLARM—The World Fish Center
M.C.A. Ablan J.W. McManus C.A. Chen K.T. Shao J. Bell A.S. Cabanban V.S. Tuan I.W. Arthana (2002) ArticleTitleMeso-scale transboundary units for the management of coral reefs in the South China Sea Area. Naga 25 4–10
S. Aris-Brosou L. Excoffier (1996) ArticleTitleThe impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol 13 494–504 Occurrence Handle1:CAS:528:DyaK28Xht1Kgurs%3D Occurrence Handle8742638
E. Arnason S. Palsson (1996) ArticleTitleMitochondrial cytochrome b DNA sequence variation of Atlantic cod, Gadus morhua, from Norway. Mol Ecol 5 715–724 Occurrence Handle1:CAS:528:DyaK2sXmtVGisg%3D%3D
S. Arnaud F. Bonhomme P. Borsa (1999) ArticleTitleMitochondrial DNA analysis of the genetic relationships among populations of scad mackerel (Decapterus macarellus, D. macrosoma, and D. russelli) in South East Asia. Mar Biol 135 699–707 Occurrence Handle10.1007/s002270050671 Occurrence Handle1:CAS:528:DC%2BD3cXnsFymtA%3D%3D
J. Avise (1998) Phylogeography. Harvard University Press Cambridge, Mass
J. Avise (2000) Phylogeography, the History and Formation of Species. Harvard University Press Cambridge, Mass
J.L. Bell J.T. Moyer K. Numachi (1982) ArticleTitleMorphological and genetic variation in Japanese populations of anemonefish Amphriprion clarkii. Mar Biol 72 99–108 Occurrence Handle10.1007/BF00396909
J.A.H. Benzie (1998) Genetic structure of marine organisms and SE Asian biogeography R. Hall J.D. Holloway (Eds) Biogeography and Geological Evolution of SE Asia. Backbuys Publisher Leiden, The Netherlands 197–209
J.A.H. Benzie (1999) ArticleTitleGenetic structure of coral reef organisms: ghosts of dispersal past. Am Zool 39 131–145
J.H. Benzie S.T. Williams (1995) ArticleTitleGene flow among giant clam (Tridacna gigas) populations does not parallel ocean circulation. Mar Biol 113 768–783
J.H. Benzie S.T. Williams (1997) ArticleTitleGenetic structure of giant clam (Tridacna maxima) populations in the West Pacific is not consistent with dispersal by present-day ocean currents. Evolution 51 768–783
G. Bertorelle M. Slatkin (1995) ArticleTitleThe number of segregating sites in expanding human populations, with implications for estimates of demographic parameters. Mol Biol Evol 12 887–892 Occurrence Handle1:CAS:528:DyaK2MXns1yqtrw%3D Occurrence Handle7476134
V.P. Buonaccorsi E. Starkey J.E. Graves (2001) ArticleTitleMitochondrial and nuclear DNA analysis of population among young-of-the-year Spanish mackerel (Scomberomorus maculatus) from the West Atlantic and Gulf of Mexico. Mar Biol 138 37–45 Occurrence Handle10.1007/s002270000439 Occurrence Handle1:CAS:528:DC%2BD3MXhsVajsbc%3D
C.A. Chen J.-K. Yu (2000) ArticleTitleUniversal primers for amplification of mitochondrial small subunit ribosomal RNA-encoding gene in scleractinian corals. Mar Biotechnol 2 146–153 Occurrence Handle10.1007/s101260000040 Occurrence Handle1:CAS:528:DC%2BD3cXjtF2jtro%3D Occurrence Handle10811953
C.A. Chen C.C. Wallace J.-K. Yu N.-V. Wei (2000) ArticleTitleStrategies for amplification by polymerase chain reaction of the complete sequence of the gene encoding nuclear large subunit ribosomal RNA in corals. Mar Biotechnol 6 558–570 Occurrence Handle10.1007/s101260000040
C.C. Cockerham (1973) ArticleTitleAnalysis of genetic frequencies. Genetics 74 679–700
P.J. Doherty P. Mather S. Planes (1994) ArticleTitleAcanthochromis polyacanthis, a fish lacking larval dispersal, has genetically differentiated populations at local and regional scales on the Great Barrier Reef. Mar Biol 121 11–21 Occurrence Handle10.1007/BF00349469
P.J. Doherty S. Planes P. Mather (1995) ArticleTitleGene flow and larval duration in seven species of fish from the Great Barrier Reef. Ecology 76 2373–2391
K.A. Donaldson R.R.J. Willison (1999) ArticleTitleAmphi-panamic germinates of snook (Percoide: Centropomidae) provide a calibration of the divergence rate in the mitochondrial control region of fishes. Mol Phylogenet Evol 13 208–213 Occurrence Handle10.1006/mpev.1999.0625 Occurrence Handle1:CAS:528:DyaK1MXmt1OmsL0%3D Occurrence Handle10508553
C.L. Dudgeon N. Gust D. Blair (2000) ArticleTitleNo apparent genetic basis to demographic difference in scarid fishes across continental shelf of the Great Barrier Reef. Mar Biol 137 1059–1066 Occurrence Handle10.1007/s002270000403
L. Excoffier (1993) MINSPNET: Minimum Spanning Network. Department of Anthropology, University of Geneva Switzerland
L. Excoffier P.E. Smouse J.M. Quattro (1992) ArticleTitleAnalysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics 13 479–491
C. Fauvelot S. Planes (2002) ArticleTitleUnderstanding origins of present-day genetic structure in marine fish: biologically or historically driven patterns? Mar Biol 141 773–788 Occurrence Handle10.1007/s00227-002-0869-7
Froese, R., and Pauly, D. (2002). FishBase. Available at http://www.fishbase.org
Y.-X. Fu (1996) ArticleTitleNew statistical tests for DNA samples from a population. Genetics 143 557–570 Occurrence Handle1:CAS:528:DyaK28XjtVKjsL0%3D Occurrence Handle8722804
Y.-X. Fu (1997) ArticleTitleStatistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 147 915–925 Occurrence Handle1:STN:280:ByiH2snjtlM%3D Occurrence Handle9335623
Y.-X. Fu W.-H. Li (1993) ArticleTitleStatistical tests of neutrality of mutation. Genetics 133 693–709 Occurrence Handle1:STN:280:ByyB3c3kvVU%3D Occurrence Handle8454210
S.W. Grant B.W. Bowen (1998) ArticleTitleShallow population histories in deep evolutionary lineages of marine fishes: insight from sardines and anchovies and lessons for conservation. J Hered 89 415–426 Occurrence Handle10.1093/jhered/89.5.415
Gilbert, D.C. (1994). SeqApp 1.9. A Biological Sequence Editor and Analysis Program for Macintosh Computers. Available at http://ftp.indiana.edu
M. Hasegawa H. Kishino T. Yano (1985) ArticleTitleDating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 21 160–174
N.L. Johnson S. Kotz (1975) Distribution in Statistics: Discrete Distributions Houghton Mifflin Boston, Mass
M. Kimura (1980) ArticleTitleA simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J Mol Evol 16 111–120 Occurrence Handle1:CAS:528:DyaL3MXmtFSktg%3D%3D Occurrence Handle7463489
W.J. Lee J. Conroy W.H. Howell T.D. Kocher (1995) ArticleTitleStructure and evolution of teleost mitochondrial control regions. J Mol Evol 41 54–66 Occurrence Handle10.1007/BF00174041 Occurrence Handle1:CAS:528:DyaK2MXmt1yktrw%3D Occurrence Handle7608989
P. Marjoram C.W. Donnelly (1994) ArticleTitlePairwise comparison of mitochondrial DNA in subdivided populations and implications for early human evolution. Genetics 136 673–683 Occurrence Handle1:CAS:528:DyaK2cXis1Kls74%3D Occurrence Handle8150290
T. Maruyama C.W. Birky (1991) ArticleTitleEffects of periodic selection on gene diversity in organelle genome and other systems with recombination. Genetics 127 449–451 Occurrence Handle1:STN:280:By6C28bivFE%3D Occurrence Handle2004715
J.W. McManus (1985) ArticleTitleMarine speciation tectonic and sea level changes in Southeast Asia. Proc 5th Intl Coral Reef Symposium (Tahiti) 133–138
J.W. McManus (1992) ArticleTitleThe Spratly Islands: a marine park? Ambio 23 181–186
J.W. McManus (1997) ArticleTitleTropical marine fisheries and the future of coral reefs: a brief review with emphasis on Southeast Asia. Coral Reefs 16 IssueIDSuppl S121–S127 Occurrence Handle10.1007/s003380050248
W.O. McMilliam S.R. Palumbi (1995) ArticleTitleConcordant evolutionary patterns among Indo-West Pacific butterfly fishes. Proc R Soc Lond B 260 229–236 Occurrence Handle7784441
J.B. Mitton W.B. Lewis (1989) ArticleTitleRelationships between genetic variability and life-history features of bony fishes. Evolution 43 1712–1723
M. Nei (1987) Molecular Evolutionary Genetics Columbia University Press New York, N.Y.
S.R. Palumbi (1992) ArticleTitleMarine speciation on a small planet. Trends Ecol Evol 112 319–326
S.R. Palumbi (1994) ArticleTitleReproductive isolation, genetic divergence, and speciation in the Sea. Annu Rev Ecol Syst 25 547–572 Occurrence Handle10.1146/annurev.es.25.110194.002555
S.R. Palumbi (1996a) Microspatial genetic structure and speciation in marine taxa with high dispersal ability. J.D. Ferraris S.R. Palumbi (Eds) Molecular Zoology Wiley-Liss New York, N.Y. 101–113
S.R. Palumbi (1996b) ArticleTitleWhat can molecular genetics contribute to marine biogeography? An urchin’s tale. J Exp Mar Biol Ecol 203 75–92 Occurrence Handle10.1016/0022-0981(96)02571-3 Occurrence Handle1:CAS:528:DyaK28XmvFOgsbk%3D
D. Pauly P. Martosubroto J. Saeger (1996) The MUTIARA 4 surveys in the Java and southern South China Sea, November 1974 to July 1976. D. Pauly P. Martosubroto (Eds) Baseline Studies in Biodiversity: The Fish Resources of Western Indonesia International Center for Living Aquatic Resources Management Manila, Philippines 47–54
N.T. Perna T.D. Kocher (1995) ArticleTitleUnequal base frequencies and the estimation of substitution rates. Mol Biol Evol 12 359–361 Occurrence Handle1:CAS:528:DyaK2MXjvVajtLs%3D
E. Petit L. Excoffier F. Mayer (1999) ArticleTitleNo evidence of bottleneck in the postglacial recolonisation of Europe by the noctule bat (Nyctalus noctula). Evolution 53 1247–1258
S. Planes C. Fauvelot (2002) ArticleTitleIsolation by distance and vicariance drive genetic structure of a coral reef fish in the Pacific Ocean. Evolution 56 378–399 Occurrence Handle1:STN:280:DC%2BD387ptlWnsw%3D%3D Occurrence Handle11926506
S. Planes P. Borsa R. Galzin F. Bonhomme (1994) Geographic structure and gene flow in the manini (convict surgeonfish, Acanthurus triostegus) in the South Central Pacific. A.R. Beaumont (Eds) Genetics and Evolution of Aquatic Organisms Chapman & Hall London, U.K 113–122
D.C. Potts (1985) ArticleTitleSea-level fluctuations and speciation in Scleractinia. Proc 5th Intl Coral Reef Symposium (Tahiti) . 127–132
M. Raymond F. Rousset (1995) ArticleTitleAn exact test for population differentiation. Evolution 49 1280–1283
W.R. Rice (1989) ArticleTitleAnalyzing tables of statistical tests. Evolution 43 223–225
C. Riginos B.C. Victor (2001) ArticleTitleLarval spatial distributions and other early life-history characteristics predict genetic differentiation in eastern Pacific blennioid fishes. Proc R Soc Lond B 268 1931–1936 Occurrence Handle10.1098/rspb.2001.1748 Occurrence Handle1:STN:280:DC%2BD3Mrhsl2mtQ%3D%3D Occurrence Handle11564350
A.R. Roger H. Harpending (1992) ArticleTitlePopulation growth makes waves in distribution of pairwise genetic differences. Mol Biol Evol 9 552–569 Occurrence Handle1:STN:280:By2B287jtFI%3D Occurrence Handle1316531
P.E. Rosel B.A. Block (1996) ArticleTitleMitochondrial control region variation and global population structure in the swordfish, Xiphias gladius. Mar Biol 125 11–22 Occurrence Handle10.1007/BF00350756 Occurrence Handle1:CAS:528:DyaK28XislKgsrY%3D
J. Rozas R. Rozas (1999) ArticleTitleDnaSP Version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15 174–175 Occurrence Handle10.1093/bioinformatics/15.2.174 Occurrence Handle1:CAS:528:DyaK1MXisVOksrY%3D Occurrence Handle10089204
S. Schneider L. Excoffier (1999) ArticleTitleEstimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152 1079–1089 Occurrence Handle1:STN:280:DyaK1MzhvVynsA%3D%3D Occurrence Handle10388826
S. Schneider D. Roessli L. Excoffier (2000) Arlequin Version 2.000: A Software for Population Genetics Data Analysis. Genetics and Biometry Laboratory, University of Geneva Switzerland
M.J. Shulman E. Bermingham (1995) ArticleTitleEarly life histories, ocean currents, and population genetics of Caribbean reef fishes. Evolution 49 897–910
G. Silvestre D. Pauly (1997) Management of tropical coastal fisheries in Asia: an overview of the key challenges and opportunities. G. Silvestre D. Pauly (Eds) Status and Management of Tropical Coastal Fisheries in Asia International Center for Living Aquatic Resources Management Manila, Philippines 8–25
M. Slatkin R.R. Hudson (1991) ArticleTitlePairwise comparison of mitochondrial DNA sequences in stable exponentially growing populations. Genetics 129 555–562 Occurrence Handle1:CAS:528:DyaK38Xhs12mtr8%3D Occurrence Handle1743491
DL. Swofford (2002) PAUP 4.0b10: Phylogenetic Analysis Using Parsimony (and Other Methods). Sinauer Associates Sunderland, Mass.
F. Tajima (1989) ArticleTitleStatistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595 Occurrence Handle1:CAS:528:DyaK3cXhslentA%3D%3D Occurrence Handle2513255
F. Tajima (1989) ArticleTitleThe effect of change in population size in DNA polymorphism. Genetics 123 597–601 Occurrence Handle1:STN:280:By%2BD1M3pslc%3D Occurrence Handle2599369
K. Tamura M. Nei (1993) ArticleTitleEstimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10 512–526 Occurrence Handle1:CAS:528:DyaK3sXks1CksL4%3D Occurrence Handle8336541
M.S. Taylor M.E. Hellberg (2003) ArticleTitleGenetic evidence for local retention of pelagic larvae in a Caribbean reef fish. Science 299 107–109 Occurrence Handle10.1126/science.1079365 Occurrence Handle1:CAS:528:DC%2BD38XpvVensL0%3D Occurrence Handle12511651
J.D. Thompson D.G. Higgins T.J. Gibson (1994) ArticleTitleCLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle1:CAS:528:DyaK2MXitlSgu74%3D Occurrence Handle7984417
J.E.V. Veron (1995) Corals in Space and Time University of New South Wales Press Sydney
B.C. Victor (1986) ArticleTitleDuration of the planktonic larval stage of one hundred species of Pacific and Atlantic wrasse (family Labridae). Mar Biol 90 317–326 Occurrence Handle10.1007/BF00428555
J.M. Waters L.H. Dijkstra G.P. Wallis (2000) ArticleTitleBiogeography of a southern hemisphere freshwater fish: how important is marine dispersal. Mol Ecol 9 1815–1821 Occurrence Handle10.1046/j.1365-294x.2000.01082.x Occurrence Handle1:STN:280:DC%2BD3M%2FosVeksA%3D%3D Occurrence Handle11091317
R.S. Waples (1998) ArticleTitleSeparating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89 438–450 Occurrence Handle10.1093/jhered/89.5.438
Acknowledgments
Many thanks to S.-Y. Rau for DNA sequencing, and C.-S. Jeng and the staff of the Penghu Aquarium, a facility of the Taiwan Fishery Research Institute, for collecting fish specimens, and K.-C. Hsui for preparing Figure 1. We thank members of the Evolution and Ecology Discussion Group, Institute of Zoology, Academia Sinica (IZAS), for constructive comments before submission, and 2 anonymous reviewers for the revision. This work was supported by grants from the John D. and Catherine T. MacArthur Foundation to the PISCES project through the WorldFish Centre, Academia Sinica Yuan Theme grants, and IZAS internal grants to C.A.C and K.T.S. This is Evolution and Ecology Group, IZAS Contribution No. 20.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, C.A., Ablan, M.C.A., McManus, J.W. et al. Population Structure and Genetic Variability of Six Bar Wrasse (Thallasoma hardwicki) in Northern South China Sea Revealed by Mitochondrial Control Region Sequences. Mar. Biotechnol. 6, 312–326 (2004). https://doi.org/10.1007/s10126-003-0028-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-003-0028-2