
Abstract
Pacific herring, Clupea pallasii, have recently colonised the northeast Atlantic and Arctic Oceans in the early Holocene. In a relatively short evolutionary time, the herring formed a community with a complex population structure. Previous genetic studies based on morphological, allozyme and mitochondrial DNA data have supported the existence of two herring subspecies from the White Sea and eastern Barents and Kara Seas (C. p. marisalbi and C. p. suworowi, respectively). However, the population structure of the White Sea herring has long been debated and remains controversial. The analyses of morphological and allozyme data have previously identified local spawning groups of herring in the White Sea, whereas mtDNA markers have not revealed any differentiation. We conducted one of the first studies of microsatellite variation for the purpose of investigating the genetic structure and relationship of Pacific herring among ten localities in the White Sea, the Barents Sea and the Kara Sea. Using classical genetic variance-based methods (hierarchical AMOVA, overall and pairwise F ST comparisons), as well as the Bayesian clustering, we infer considerable genetic diversity and population structure in herring at ten microsatellite loci. Genetic differentiation was the most pronounced between the White Sea (C. p. marisalbi) versus the Barents and Kara seas (Chesha–Pechora herring, C. p. suworowi). While microsatellite variation in all C. pallasii was considerable, genetic diversity was significantly lower in C. p. suworowi, than in C. p. marisalbi. Also, tests of genetic differentiation were indicating significant differentiation within the White Sea herring between sympatric summer- and spring-spawning groups, in comparison with genetic homogeneity of the Chesha–Pechora herring.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pacific herring, Clupea pallasii, have an interrupted distribution. They are widely distributed over large areas on both sides of the North Pacific Ocean and also inhabit the North European Seas of the Atlantic Ocean and Arctic region: from the White Sea eastward to the southern part of the Kara Sea. Isolated small populations may occur in the estuarine waters of large Siberian Rivers and some Norwegian Sea fjords (Jørstad et al. 2001). Three subspecies have been recognised: nominal Pacific herring C. p. pallasii Valenciennes, 1847; the White Sea herring C. p. marisalbi Berg, 1923; and the Chesha–Pechora (Kanin–Pechora) herring C. p. suworowi Rabinerson, 1927 in the south-eastern Barents and southern Kara Sea (Svetovidov 1952).
The White Sea and, especially, the southern Barents and Kara seas are geographically peripheral parts of Arctic Ocean. As such they are characterised by severe climatic conditions including several months of sea ice cover each year. The climate in the White Sea has pronounced continental features, with relatively warm summers and long severe winters. Ice covers the bays of the sea from December until May (Berger and Naumov 2001). Ice cover in the Barents and Kara seas is formed annually, lasting for 8 months from October until June. The Barents Sea is influenced by warm and saline Atlantic waters, while the Kara Sea is almost closed off from the Atlantic (Kulakov et al. 2006). Recent glaciations have resulted in major losses of the resident fauna, and recolonisation has been slow, owing to both the extreme environmental conditions and the low overall productivity of the Arctic ecosystem (ACIA 2004).
The herring colonisation of northern Europe resulted from invasion through the Arctic from the Pacific Northwest in the early Holocene, since the opening of the Bering Strait (<12 kya) (Laakkonen et al. 2013). At present, the greatest abundance of Pacific herring in this area is observed in the White Sea and the south-eastern Barents Sea. These populations of C. pallasii display considerable variation in life history. The analyses of morphometric, spawning time, locations and growth patterns have shown differences between herring stocks. Thus, the inner White Sea herring have differentiated into several geographical and temporal spawning stocks, whose population status has long been debated (Lajus 2002). The most striking differences have been revealed between two sympatric groups: fast-growing summer-spawning herring of the open sea (Kandalaksha Bay and Solovetsk Island) versus slow-growing spring-spawning herring of coastal waters from the Kandalaksha, Onega and Dvina bays, which breed under the ice. The summer-spawning herring attain 34 cm, maturing at 3–4 years of age. They spawn in June at depths down to 5 m, at 5–15 °C, with an age limit of 13–14 years. The spring-spawning herring rarely attain 20 cm, maturing at the age of 2–3 years. The spring spawners form distinct breeding stocks, which are recognisable from their spawning grounds and spawning time, as well as morpho-biological traits, such as growth rate or age of maturity (Altukhov 1958). The southeast Barents Sea and the Kara Sea are occupied by a different form of herring, called by Svetovidov (1952) the “Chesha–Pechora”. Chesha–Pechora herring lengths reach up to 32 cm, and their lifespan is 11–12 years; they mature at the age of 4–5 years. Herring stocks spawn from mid-May until July in Chesha Bay and in August–September in the Kara Sea.
The current understanding of the genetic population structure of herring is fragmentary, and there is no general agreement as to how herring populations are spatially and temporally structured. Allozyme data have revealed three genetic groups from the survey area: White Sea spring-spawning herring, White Sea summer-spawning herring and Chesha–Pechora spring-spawning herring (Semenova et al. 2004, 2009). Significant heterogeneity among sympatric seasonal stocks of herring in the White Sea was also detected by microsatellite analyses (Semenova et al. 2013). In contrast, the data of chromosomal sets and the variation of the meristic characters revealed significant polymorphism between the major bays of the White Sea, but not between sympatric seasonal breeding stocks (Lajus 1996, 2002). The mtDNA data failed to demonstrate differences, either between the main spawning regions in the inner White Sea or between the spring- and summer-breeding cohorts. However, they have shown differentiation among the White and Barents seas’ herring (Laakkonen et al. 2013). The two different classes of molecular markers (mitochondrial and microsatellite) may be reflecting different and complementary aspects of the evolutionary history of herring. The mtDNA genetic variation is less helpful when tackling questions about present day genetic structuring of taxa with large population sizes and high levels of gene flow within their distribution such as marine pelagic fishes (Hauser and Ward 1998; Goldstein et al. 1999; Buonaccorsi et al. 2001; Ruzzante et al. 2006).
The economic importance of herring clearly emphasises the need for a better understanding of the genetic population structure of the species, as well as the mechanisms underlying its population dynamics. The goal of the present study was to describe the population structure and variability of Pacific herring over the entire distributional range in the North European seas. This is the first microsatellite DNA-based population genetic analysis comparing Pacific herring sampled in the White Sea and the Chesha–Pechora region. We investigated the genetic relationship of herring with two types of genetic markers: gene-associated and anonymous microsatellite markers. The anonymous microsatellites are widely used for detecting fine-scale population structures in marine pelagic fishes, including Pacific herring (Sunnuks 2000; Durand et al. 2005; Wildes et al. 2011). Also, we took advantage of newly developed gene-associated microsatellites, which may provide a perspective in stock discrimination studies (Shikano et al. 2010; Teacher et al. 2011) and compared the patterns of population differentiation revealed by two types of markers. The results are considered in relation to the northeast Atlantic C. pallasii herring, life-history variation and population history, and an attempt is made to reconcile the current views of the herring population structure. Finally, we conclude by discussing the implications of our findings for the management of herring fisheries in the White Sea and southern Barents Sea.
Materials and methods
Sampling
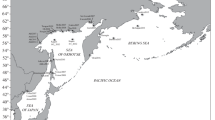
In this study, ten samples of C. pallasii from the White Sea, Barents and Kara seas (Chesha–Pechora region) were analysed. These samples were collected in 2000–2008 (Table 1; Fig. 1). The samples from the Kara Sea (KAR), the Barents Sea (CHS1, CHS2, IND) and the White Sea (ONG, CHP) were spring-spawning herring. The sample RUG from the Rugozero Inlet (Kandalaksha Bay, White Sea) was a summer-spawning herring. The sample from the Barents Sea, Vaygach Island (VAY), was taken in October and not during the spawning season. Two samples from the Solovetsk Island, White Sea (SLS and SLB), were non-spawning. However, we referred them to the summer-spawning group due to their morpho-biological traits. Therefore, the sample SLS from the Sosnovaya Inlet consisted mostly of individuals that were at stage IV of gonad maturity (pre-spawning conditions). The herring sample SLB from the Blagopoluchiya Bay was collected after the spawning period and had a gonad maturity stage of VI-II. All samples could be classified by phenotype; herring from the Solovetsk Islands (SLS and SLB) and from Rugozero Inlet (RUG) were all large, fast-growing summer-spawning herring. All fish sampled in the White Sea (ONG, CHP) were small, slow-growing spring-spawning herring (for further details, see Semenova et al. 2004, 2009, 2013).

Sampling locations for Pacific herring. Populations are marked by abbreviations that correspond to Table 1
Microsatellite analyses
DNA was extracted from tissue samples (muscle, fin or scale) by the following standard methods using the Diatom DNA Prep kits (IsoGene, Russia). PCR amplification was performed using the GenePak PCR Core kits. The reactions were run in a final volume of 20 μl including the incubation mixture of 10 μl PCR buffer, 200 μl of each dNTP, 1.5 mM MgCl2, 50 ng DNA, and 100 ng of a specific primer. PCR were conducted on an MJ Research PTC-100 thermal cycler. The reaction conditions included denaturation at 94 °C for 2 min, followed by eight cycles of denaturation at 94 °C for 1 min; a primer-specific annealing at 52 °C for 30 s; and extension at 72 °C for 30 s. This was followed by 21 cycles at 94 °C for 30 s; at 52 °C for 30 s; at 72 °C for 15 s; and a final extension at 72 °C for 3 min.
In total, we tested eight genome-derived and ten transcriptome-derived microsatellite loci that were originally designed for herring by Olsen et al. (2002), Miller et al. (2001), McPherson et al. (2001) and Teacher et al. (2011). Eight of them were excluded from the data analyses due to poor amplification success or the fact that they proved to be monomorphic. Finally, ten tetra-nucleotide microsatellite loci were genotyped, consisting of five transcriptome-derived loci (Teacher et al. 2011): Her18, Her28, Her36, Her71 and Her140; and five genome-derived loci (McPherson et al. 2001; Miller et al. 2001; Olsen et al. 2002 ): Cpa110, Cpa113, Cpa4, Cha1059 and Cha1020.
Amplification products were separated by means of electrophoresis in 6 % non-denaturing polyacrylamide gels in 0.5×TBE buffer at 300 V for 2–3 h. The gels were stained with ethidium bromide and photographed under UV light. DNA of the pBr322 plasmid, digested with either HaeIII or HpaII restriction endonucleases, was used as a molecular size marker. The allele sizes at each locus were determined in accordance with the electrophoresis mobility of the band using the 1D Image Analysis Software Version 3.5 program (Kodak).
Descriptive genetic data
Samples were tested for deviation from HW equilibrium and linkage disequilibrium by using GENEPOP 3.4. (Raymond and Rousset 1995). The data were examined for all possible scoring errors, null alleles or large allele drop-out with the MICROCHECKER v.2.2.3 (van Oosterhout et al. 2004). Departures from neutrality of loci were tested using LOSITAN (Antao et al. 2008) with 100,000 simulations, “neutral” mean F ST, confidence intervals of 95 % and a false discovery rate (FDR) of 0.05 with both the infinite alleles (IAM) and the stepwise mutation (SMM) models. We conducted a global outlier test using all of the populations and pairwise tests between both subspecies.
We used POWSIM 4.1 (Ryman et al. 2006) to estimate statistical power in order to detect various levels of differentiation with the ten microsatellites. χ 2 and Fisher’s probabilities were used to test a range of predefined levels of expected divergence F ST. Divergence among populations was modelled by allowing frequencies to drift for a given number of generations with effective population sizes N e = 2,000. Simulations using our empirical microsatellite data for all loci dataset and for genome-derived and transcriptome-derived loci separately, taking into account the sample size for each population and a wide range of predefined F ST values, showed that total marker sets have enough statistical power to detect F ST > 0.005 with a probability close to one (Online Resource 1). At lower levels of divergence, the genome-derived loci are more powerful for detecting structuring than the transcriptome-derived loci.
The GDA 1.0 (Lewis and Zaykin 2001) was used for calculating allele frequencies, the expected and observed heterozygosities (H E, H O), the allelic diversity (A) and the number of private alleles (A PR). The allelic richness (A R), corrected for the minimum sample size, was calculated using FSTAT 2.9.3 (Goudet 2001). A one-way ANOVA was performed using the add-in “Analysis ToolPak” in Excel to test for differences in the A R and the H E between sites.
We performed (using Excel) two-sample t tests (unpaired, not assuming equal variance, two-tailed) to estimate differences between the mean locus-specific F ST, H E and A R of each marker type: transcriptome- and genome-derived.
In order to detect indications for a recent bottleneck or a reduction in population size, we used BOTTLENECK 1.2.02 (Piry et al. 1999). We performed the one-tailed Wilcoxon signed-rank test with the two-phase model (TPM) and SMM; 1,000 iterations were applied. We selected the TPM method with the program’s default parameters: 30 % single-step and 70 % multiple-step mutations. A significant heterozygosity excess indicates a recent bottleneck event (Cornuet and Luikart 1996). For each population, only polymorphic loci in HWE were used.
Population differentiation analyses
Population differentiation was estimated per sample pair and overall using F ST or θ (Weir and Cockerham 1984) in GENEPOP 3.4. or GDA 1.0, respectively. Fisher’s exact test in GENEPOP was used to test for significant differences in allele frequencies between populations. We compared the level of variation among samples based on allele identity (F ST) with the level of variation based on allele size (R ST). We carried out a permutation test (1,000 permutations) in order to test the null hypotheses of no existing contribution of stepwise mutation to genetic differentiation (R ST = F ST) using SPAGEDi 1.2 (Hardy and Vekemans 2002). All probability tests were based on the Markov chain method (Guo and Thompson 1992; Raymond and Rousset 1995) by using 1,000 steps, 100 batches and 1,000 iterations per batch. The sequential Bonferroni adjustments (Rice 1989) were applied to correct for the effect of multiple tests.
A Mantel test, employing geographical distances between populations (shortest marine distance, km) and F ST estimates, was used to test for an isolation-by-distance relationship between populations using IBDWS 3.23 (Jensen et al. 2005). The probability that a correlation was significantly larger than zero was tested with 1,000 randomisations. Log transformations of the variables were made in an attempt to improve the correlation.
The Bayesian clustering method implemented in STRUCTURE 2.3.4 (Pritchard et al. 2000) was used to assign individuals to clusters using no prior information on what sites the individuals belong to. Considering previous findings of high levels of gene flow in herring, we used the admixture model with correlated allele frequencies to reflect the most likely pattern of population connectivity. STRUCTURE was run using MCMC simulations of 800,000 iterations, with a burn-in period of 400,000. Runs were conducted for the number of putative populations (i.e. K), set at 1–10, with each having three iterations. The result files were then used in Structure Harvester (Earl and vonHoldt 2012) in order to estimate the uppermost optimal number of clusters using Evanno’s ∆K method (Evanno et al. 2005).
Evanno et al. (2005) suggests that STRUCTURE only captured the major structure in the data, although a subsequent analysis performed on each identified cluster can potentially demonstrate a more intricate population structure within these clusters. This study used the “hierarchical STRUCTURE analysis” as outlined by Vähä et al. (2007), Warnock et al. (2010) and Olafsson et al. (2014). Calculation was made with 500,000 burn-in period and 106 MCMC with K ranging from one to five, with three iterations per each K. STRUCTURE was run independently both with and without sampling location information. The estimated number of group (K) was based on ∆K and changes in the pattern of LnP(D) values.
The hierarchical analyses of molecular variance (AMOVA) were calculated using ARLEQUIN 3.11. (Excoffier et al. 2005) with 10,000 iterations. Because earlier investigations based on allozyme data (Semenova et al. 2009) had indicated that spring-spawning and summer-spawning White Sea herring were both significantly different from each other and from the Chesha–Pechora herring, we conducted the AMOVA with these three groups. Also, the hierarchical structure was estimated with only two groups of herring: the White Sea, C. p. marisalbi versus the Barents and Kara seas herring, C. p. suworowi.
The BARRIER 2.2 (Manni et al. 2004) was used to highlight the geographical areas with pronounced genetic discontinuity between the samples, i.e. barriers. Geographical coordinates were supplied for each sample and connected by Delauney triangulation, such that each connection had an associated distance (in this case, the genetic distance F ST described above). Using Monmonier’s maximum distance algorithm, barriers were then identified. In short, this algorithm chooses the edge with the largest associated distance in the triangulation network as its starting edge and then extends the barrier across the directly adjacent edge associated with the largest genetic distance. Additional sections are added to the barrier by such steps until the barrier reaches the outer edge of the network or meets another barrier (Manni et al. 2004).
Effective population size and gene flow
Long-term gene flow (M = m/μ, where m = migration rate and μ = mutation rate) and θ, a measure of effective population size (θ = 4N e μ; where N e = effective population size, and μ = mutation rate), were estimated with a coalescent method by using the Bayesian implementation in MIGRATE 3.2.1 (Beerli 2003). We estimated gene flow among three clusters that were previously identified by STRUCTURE. The microsatellite mutation was modelled as a continuous Brownian process and was allowed to have different rates among the loci. For each run, we used ten short chains of 50,000 sampled and 500 recorded trees, followed by three long chains of 500,000 sampled and 5,000 recorded trees. An adaptive heating scheme using four simultaneous Markov chains was implemented in order to increase the search efficiency. Initial estimates of θ and N e m were generated from F ST values. The asymmetric gene flow is confirmed by a one-way ANOVA applied to test the null hypothesis of no differences in the immigration rates among the clusters (P < 0.01).
Results
Microsatellite diversity and tests for bottleneck
A total of 105 alleles were observed across the ten microsatellite loci, with an average of 10.5 alleles per locus. They range from three at Her18 to 22 at Cha1020 (Table 2). The levels of average heterozygosity and allelic richness per collection ranged from 0.405 to 0.593 and from 3.36 to 5.63, respectively. The one-way ANOVA showed that the A R and H E did not differ significantly between sites (A R: F = 0.333, P = 0.961; H E: F = 0.277, P = 0.979).
The H E and A R were significantly lower in the transcriptome-derived loci than in the genome-derived loci (H E transcriptome = 0.304; H E genome = 0.782, t = −19.8, P < 0.001; A R transcriptome = 2.58; A R genome = 7.04, t = −13.01, P < 0.001).
The diversity estimates were lower in the Barents and Kara Seas herring (H E = 0.405–0.510; A R = 2.57) than in the White Sea herring (H E = 0.515–0.593; A R = 2.84). T test values were significant for both the H E (t = 3.16, P = 0.006) and the A R (t = 3.57, P = 0.003).
There were no indications of bottlenecks in any of the samples for the TPM (0.898 < P<0.994) or the SMM (0.981 < P < 1.000).
Hardy–Weinberg equilibrium and linkage disequilibrium
Of the 100 single-locus tests for conformation to HWE, 11 had significance values of P <0.05, but only two (Cha1059, CHP and KAR) tests showed significant departure from the expectations following Bonferroni corrections (Table 2). When the results of all loci were combined for each population, only CHP departed from HWE after correction (P = 0.0001). In LD tests within samples, ten of the 485 tests were significant before correction, but none of the tests were significant after correction.
Data quality check and outlier tests
MICROCHECKER suggested a possible null allele at Cha1059 in three samples: KAR, CHP and RUG. No other loci showed signs of possible null alleles. There was no evidence for scoring error due to shuttering or large allele drop-out for any microsatellite loci. The locus Cha1059 did not appear to be an outlier in other respects; however, we performed further analyses both with and without Cha1059 to assess the effect of the locus.
The global LOSITAN analysis under the SMM for all samples detected significant departures for Cha1020, which indicates a putative balancing selection (Online Resource 2). While there were no significant selection influences for any of the loci under the IAM. The pairwise outlier analysis between subspecies founded no evidence for selection under SMM and IAM models. Nevertheless, we performed further analyses both with and without Cha1020.
Genetic diversity within C. p. suworowi
The genetic homogeneities were observed within the Barents and Kara Seas samples (CHS1, CHS2, IND, VAY and KAR), based on the F ST and R ST pairwise estimates (Exact tests, P = 0.032–0.443) (Table 3). The overall θ was also not significant θ = 0.019, CI −0.001 to 0.033. A re-analysis of the data, excluding the Cha1020 and Cha1059 loci, provided approximately the same θ = 0.017 CI −0.001 to 0.037, t = −0.455, P = 0.655.
Genetic differentiation did not follow an isolation-by-distance model for the Chesha–Pechora herring (Mantel tests: all loci: r = −0.294, P = 0.740; excluding Cha1059 and Cha1020: r = −0.259, P = 0.617) (Online Resource 3ab).
Genetic diversity within C. p. marisalbi
A pairwise population analysis of White Sea herring (RUG, ONG, CHP, SLS and SLB) detected a significant difference in allele frequencies for nine of the ten possible tests (Exact tests, P = 0.000–0.0005), with multilocus F ST ranging from −0.001 to 0.040. The genetic differentiation was not significant only between SLS versus SLB. Pairwise R ST was significant for RUG vs. ONG and CHP, but all values were non-significant after correcting for multiple testing (Table 3).
The overall θ = 0.020, CI 0.009–0.034 was significant, and it remained similar after the removal of the Cpa1020 and Cha1059 loci (θ = 0.025 CI 0.016–0.042, t = −0.03, P = 0.971).
Tests for IBD were not significant for C. p. marisalbi (all loci: r = 0.804, P = 0.122; excluding Cha1059 and Cha1020: r = 0.805, P = 0.130) (Online resource 3cd).
However, a hierarchical AMOVA revealed significant genetic variation within the White Sea with two a priori clusters: spring- and summer-spawning herring (Table 4). Genetic variation between sympatric spawners explained 1.43 % of all microsatellite variation observed.
Population genetic structure of C. pallasii
The overall θ = 0.034, CI 0.020–0.048 revealed a significant level of genetic structuring among all samples investigated. Individual locus estimates of θ varied considerably, nine associated exact tests of global differentiation were significant (P < 0.05). There was no significant differentiation at only Her18 locus (Online Resource 4). The value of genetic differences was slightly higher (θ = 0.042 CI 0.027–0.055) after the removal of the Cpa1020 and Cha1059 loci, but was not significant with the θ of all loci datasets (t = −0.421, P = 0.679).
All pairwise F ST, and eight from the 25 pairwise R ST, were significant in C. p. suworowi versus C. p. marisabli (Table 3). The allele size permutation test found non-significant differences between all F ST and R ST estimates (P = 0.094) and supported the existence of a population structure within C. pallasii, probably due to genetic drift and not to mutation.
A significant correlation between the genetic and geographical distances appeared across the entire geographical range (Mantel test; all loci: r = 0.472, P = 0.009; excluding Cha1059 and Cha1020: r = 0.476, P = 0.007) (Online resource 3ef).
As the removal of the Cha1059 and Cha1020 loci made no significant difference in any of the statistical values, we will concentrate on further analysing all loci datasets.
The Bayesian clustering analyses using STRUCTURE suggested that the largest ∆K appeared under the assumption of three groups (Online Resource 5). Most individual genotypes indicated admixture between clusters, but, overall, the three identified clusters corresponded with: (1) the Barents Sea and the Kara Sea (CHS1, CHS2, IND, VAY, KAR), (2) the White Sea summer-spawning herring (RUG, SLS, SLB) and (3) the White Sea spring-spawning herring (CHP and ONG) (Online Resource 6A). A model of K = 2 was also statistically most likely. Here, two clusters corresponded with C. p. marisalbi (the White Sea) versus C. p. suworowi (the Barents and Kara seas) (Online Resource 6B). The second round of hierarchical STRUCTURE analysis was subsequently carried out on each of two subspecies’ clusters separately. The White Sea initial cluster was separated into the two previously described groups of sympatric spawning herring (K = 2), while no subdivision was indicated within the Barents and Kara seas initial cluster (both with and without location information). The third round of STRUCTURE analysis has not identified any clusters within spring-spawning or summer-spawning groups, running the model without prior information on sampling location. Whereas the results based on the STRUCTURE model incorporating the LOCPRIOR indicated further clustering within these sympatric groups. Populations detected at this level were RUG (Kandalaksha Bay) and SLS/SLB (Onega Bay) groups within the summer-spawning cluster and CHP (Kandalaksha Bay) and ONG (Onega Bay) groups within the spring-spawning cluster.
The results of the hierarchical gene diversity analysis revealed that the two gene pool structures separating the subspecies C. p. marisalbi versus C. p. suworowi were significant (P < 0.001) (Table 4). The AMOVA showed that 3.59 % of the variation was due to differences between the two subspecies and 1.52 % for differences within subspecies. A possible hypothesis of structuring into three groups was also significant in AMOVA: 3.51 % of the total genetic variation was accounted for by differences between the three Bayesian clusters [spring-spawning (White Sea), summer-spawning (White Sea) and spring-spawning (Barents and Kara seas)], 0.78 % was due to differentiation among the spawning localities within the sympatric cohorts, and 95.7 % was due to variation in the sample collections. All levels of genetic differences were significant (P < 0.001).
Based on pairwise F ST, the barrier analyses identified two areas of relatively sharp changes in genetic composition. The first barrier (a) separated the White Sea herring from the Chesha–Pechora herring. The second barrier (b) was located within the White Sea and, separated the CHP (Chupa Inlet, Kandalaksha Bay) sample from the sample RUG (Rugozero Inlet, Kandalaksha Bay) on one hand, and samples SLS and SLB from the Solovetsk Island, Onega Bay, on the other (Online Resource 7). It should be noted that the second barrier reflects diversity in spawn timing between the White Sea herring: herring from the ONG are spring-spawning while herring in samples RUG, SLS and SLB are summer-spawning.
Effective population size and gene flow
The estimated population size parameters and migration values of herring are shown in Table 5. The long-term effective population size (N e) was obtained by assuming the average mutation rate of the microsatellites to be 10−4 mutation/generation/locus (Whittaker et al. 2003). Estimates of N e in summer-spawning C. p. marisalbi were smaller (45 individuals) than were those in the spring-spawning C. p. marisalbi and C. p. suworowi (245 individuals). These N e estimates are relatively small, compared with the estimated census population size (as derived from catch data, about 10,000 tonnes for White Sea herring). Conspicuously, small N e/N ratios are commonplace in marine fishes, including herring, and can be explained either by historical events such as past population bottlenecks or by life-history traits such as strong bias in reproductive success or size-dependent fecundity (Hedgecock 1994; Hauser et al. 2002; Shrimpton and Health 2003; Gonzalez et al. 2008; Larsson et al. 2010; Palstra and Fraser 2012).
Migration rates between the three groups were symmetric. The one-way ANOVA showed no significant variation of the number of immigrants among the clusters (F = 1.69, P = 0.321). Inferred numbers of immigrants per generation (N m = θ × M/4) between groups ranged from 0.22 to 2.86. These estimates of migration rates were smaller than N m for other pelagic fishes, such as sardines (Gonzalez and Zardoya 2007).
However, our estimates of population size parameters and gene flow were obtained on a non-homogenous sample size collected over a long time span, so the results of this analysis have to be taken with caution. In addition, the actual microsatellite mutation rate in herring is not known.
Discussion
This study has revealed considerable genetic diversity and population differentiation in the herring species C. pallasii from the northeast Atlantic. Genetic differentiation was most pronounced between the White Sea (C. p. marisalbi) versus the Barents and Kara Seas (Chesha–Pechora herring, C. p. suworowi), indicating that populations in these two areas are relatively isolated. We found the existence of a barrier to gene flow between the White Sea and southern Barents Sea herrings. While microsatellite variation in all C. pallasii was considerable, genetic diversity was significantly lower in C. p. suworowi, than in C. p. marisalbi. Tests of genetic differentiation were also significant indicating significant differentiation within White Sea herring between sympatric summer- and spring-spawning groups, in comparison with the genetic homogeneity of the Chesha–Pechora herring.
Before discussing these findings in detail, we mention that even though the sample size in some samples was small, these samples sizes were still large enough and the microsatellite marker polymorphic enough to provide power to detect population structure as low as F ST = 0.005 about 99 % of the time (Online resource 1). This level of divergence is much smaller that the level of divergence within C. p. suworowi and C. p. marisalbi.
Microsatellite diversity and bottleneck events
The Pacific herring populations surveyed in this study revealed medium levels of allelic diversity, H E = 0.512. The value of diversity estimated on only genome-derived loci H Egenome = 0.744 was slightly less than was the microsatellite-based heterozygosity reported for Pacific herring: H E = 0.815–0.900 (O’Connell et al. 1998; Beacham et al. 2002; Small et al. 2005; Sugaya et al. 2008), but on average it is similar to that recently reported in NW and NE Pacific herring with an H E = 0.672–0.811 (Liu et al. 2012).
The lower genetic diversity may reflect population bottlenecks from environmental variability at the edge of the species’ distribution and physical properties of the environment, such as geographical or ecological periphery (Lesica and Allendorf 1995; Schwartz et al. 2003; Johannesson and André 2006; Liu et al. 2012; Teacher et al. 2013).
The lower values of genetic diversity demonstrated in C. p. suworowi compared with C. p. marisalbi could be explained by differences in the spatial heterogeneity between the White Sea and southern Barents and Kara Seas. It is generally accepted that greater environmental heterogeneity will maintain a higher level of genetic variation within the species (Hedrick 1986). The White Sea has high heterogeneity in herring habitats, due to being located in semi-isolated bays and open sea water. These areas significantly differ from each other in their physical properties, climatic characterisation, system of tides, salinity etc. (Pantyulin 2003). At the present time, the environmental conditions of the Chesha–Pechora region and, especially of the Kara Sea, are more severe than those of the White Sea (Matishov et al. 2004; Kulakov et al. 2006). A likely explanation for the lower levels of genetic variation in the southern Barents Sea and Kara Sea is that population oscillation in herring from this area, especially from the Kara Sea, can lead to the stochastic loss of genetic variability and bottleneck events.
In addition, the Arctic seas have only recently been available for the population of Pacific herring (from 12 to 5–8 kya). The species from the newly colonised areas have significantly lower genetic diversity than does the parent population (Hewitt 1996; Grant and Bowen 1998; Limborg et al. 2009; Glover et al. 2011). Therefore, Pacific herring populations from the White Sea, eastern Barents Sea and Norwegian Sea (Balsfjord) showed reduced mtDNA variation. It is possible that these populations have lost diversity in relatively recent colonisation bottlenecks (Laakkonen et al. 2013).
However, there are no indications of bottlenecks in our microsatellite data for any of the herring samples. Probably, the most likely explanation is that populations have lost substantially more genetic diversity of mitochondrial genes compared with nuclear genes (Avise 1994). Moreover, microsatellite-based bottleneck tests can have limited statistical power to detect bottlenecks largely as a result of limited sample sizes (Peery et al. 2012).
At the same time, our long-term N e estimates are small and can be explained by historical population bottlenecks (Hauser et al. 2002).
Population structure of C. pallasii
The most significant finding of this study was the high degree of population differentiation of C. pallasii in the northeast Atlantic. The level of differentiation was surprising given the small geographical separation of approximately 1,000 km between the two outermost sample localities (less than 400 km within the White Sea samples), as well as the higher genetic connectivity that is typical among marine pelagic species, such as herring (Ward et al. 1994; Hauser and Carvalho 2008).
The overall θ = 0.034 was larger than were the typical estimates uncovered in previous studies of microsatellites in herring over much larger geographical areas. For example, the mean level of F ST ranged from 0.001 to 0.003 within NE and NW Pacific herring (Beacham et al. 2002; Small et al. 2005; Sugaya et al. 2008; Wildes et al. 2011) and up to 0.036 (O’Connell et al. 1998). Moreover, the same microsatellite loci analyses have not shown significant genetic differentiation among spawning groups of Pacific herring from the Sea of Okhotsk (Semenova et al. 2014).
The Bayesian approach of STRUCTURE detected C. p. marisalbi and C. p. suworowi in different clusters. Genetic variation between these subspecies groups explained 3.59 % of all of the microsatellite variation observed. We also found the existence of a barrier to gene flow between the White Sea and southern Barents Sea herrings. Together with the reduced allelic diversity observed in all Chesha–Pechora herring, it is our opinion that the data indicate limited connectivity among these subspecies groups.
Large-scale genetic differentiation between C. p. suworowi and C. p. marisalbi correlated with the geographical distance between the populations. However, with the result of this analysis, it is difficult to make an inference regarding migration-drift equilibrium, because a false-positive signal of isolation by distance can occur when divergent populations are included in the analysis and the Barrier effects can contribute a stronger IBD patterns (Hutchinson and Templeton 1999; Koizumi et al. 2006).
The divergence between herring populations may have initially evolved as a result of physical characteristics of the White Sea. The three regions, Voronka (funnell), Mezen Bay and Gorlo (throat) strait, form the northern part of the White Sea; the southern part is the most individual part of the sea, which includes the Basin and three bays (Dvina, Onega and Kandalaksha) (Pantyulin 2003). Although the boundary between the Barents Sea and the White Sea goes along the line from Cape Kanin Nos to Cape Svyatoi Nos, the strong stream in the long (50 km), narrow and shallow Gorlo strait potentially acts as a barrier to dispersal between populations.
Unfortunately, we have no microsatellite data about herring from the Mezen Bay, but earlier studies found that Mezen herring, formally belonging to the White Sea, are very close in morpho-biological traits, allozyme and mtDNA markers to the Chesha–Pechora herring (Dmitriev 1946; Svetovidov 1952; Altukhov 1958; Semenova et al. 2009; Laakkonen et al. 2013). The physical barriers to gene exchange can result in population structuring of herring in this region.
The non-significant comparisons of the pairwise estimates of F ST and R ST suggest that the two subspecies diverged relatively recently. If this diverging was recent, the differences between the populations would be due primarily to genetic drift and not to mutation. As a result, the estimates of F ST would be similar to the estimates of R ST. Alternatively, if populations historically diverged from one another, differentiation would also be the result of stepwise mutations, and R ST would be greater than F ST (Hardy et al. 2003). Estoup and Angers (1998) argue that mutations are expected to have a substantial effect on population divergence only after 2,000 generations assuming a typical rate of mutation for the microsatellites. Generation lengths for herring are usually about 2–4 years, which can be transformed in no more than 8,000 years of interspecies divergence.
These results are congruent with those derived from the analyses based on mtDNA, which have shown differentiation among the White and Barents seas’ herring (Laakkonen et al. 2013). The authors concluded that the complex substructure of C. pallasii arose during regional refugial phases associated with post-glacial climatic fluctuations. Genetic drift and limited migration must have been the primary factors in generating the structure, while selection that restricts gene exchange between locally adapted stocks might also have contributed to its maintenance.
The primary role of subspecies differences between C. p. marisalbi versus C. p. suworowi in our opinion is spatial isolation and limited connectivity among herring groups due to physical barriers to the dispersal of larvae, as well as to adult herring appearing in the Gorlo strait.
Genetic diversity within C. p. suworowi and C. p. marisalbi
The genetic relationships among the sites within the subspecies groups could not be explained by their geographical location. Neither the White Sea herring, nor the Chesha–Pechora/Kara Sea herring are structured according to the isolation-by-distance model.
Within the Barents and Kara seas no significant genetic differentiation between geographical locations was observed (hierarchical STRUCTURE analysis, overall θ, all F ST and R ST pairwise comparisons rendered low and non-significant values). These results support unrestricted gene flow among C. p. suworowi.
In contrast, gene flow in C. p. marisalbi is limited across relatively small spatial scales. Separate analyses of samples within the White Sea revealed significant structuring of genetic variation. The observed pattern may be due, in part, to isolation spawning time differences among populations. The summer-spawning herring (RUG, SLS and SLB) and spring-spawning (ONG, CHP) formed separated Bayesian clusters in STRUCTURE. The genetic differentiation between them explained 1.43 % of all variation observed in C. p. marisalbi. Moreover, areas of lowered gene flow were identified by BARRIER, leaving two groups of samples: spring-spawning (CHP, Kandalaksha Bay) and summer-spawning (RUG, Kandalaksha Bay; SLS and SLB, Onega Bay) herring. The migration rates (numbers of immigrants per generation N m) in summer-spawning herring did not exceed one, as estimated by MIGRATE, and cannot counteract genetic drift and population differentiation (Mills and Allendorf 1996). This finding is not entirely unexpected, because the White Sea herring species is known to exhibit substantial morphological and life-history differentiation (Lajus 2002). Molecular genetic differences between the spawning groups employing allozyme and microsatellite DNA coincide with the morphological and ecological differences between them. Spring-spawning herring are generally smaller, grow more slowly and mature earlier than do summer-spawning herring. Spatial differences in growth indicate the different ecology of these groups and, obviously, a different life strategy. The spring-spawning herring occupy coastal areas, feed mainly on small zooplankton and are mature rapidly. In contrast, the summer-spawning herring occupy the open sea, make migrations, feed on large zooplankton and attain maturity 2–3 years later (Semenova et al. 2009).
MIGRATE results showed that the effective size of spring-spawning herring is five times larger than that of summer-spawning herring. This can be explained by the quantity of occupied spawning grounds. The spring-spawning herring are widely distributed in all of the bays in the White Sea, while summer-spawners breed only in Kandalaksha Bay and near the Solovetsk Island.
Moreover, the results of hierarchical STRUCTURE analysis indicated the existence of further sub-structuring of both summer-spawning and spring-spawning groups of herring. At least two subpopulations from different bays were detected within each of the sympatric group. However, as it is not possible to assess the detailed population structure within summer- and spring- spawning groups due to insufficient data, more complex studies are warranted to further investigate this issue.
Genetic differences of C. p. marisalbi can be maintained by specific hydrological characteristics of the White Sea that promote the restriction of larvae dispersion from the spawning grounds. Based on long-term investigations of the dispersal of herring larvae with the involvement of the up-to-date data on hydrological structure of the White Sea waters, the absence of larval exchange between the herring stock spawning in Kandalaksha, Onega and Dvina bays was demonstrated (Pantyulin 2003; Evseenko et al. 2006; Mishin et al. 2008; Evseenko and Mishin 2011). The retention of larvae in the shallow waters of the Kandalaksha Bay is due to the double-layer water circulation system that exists in the regions of herring spawning. The export of larvae outside the shallow waters of the Onega and Dvina bays can be limited by frontal water parting, which exists at the borders between these bays and the White Sea Basin. It is also suggested that juvenile herring remain close to the spawning grounds (Evseenko and Mishin 2011). Despite the fact that herring can spread over the area of the White Sea, where the mixing of local stocks occurs for the purpose of feeding, in autumn, they return to the bay inlets and likely move to their spawning grounds to spawn (Altukhov 1958). Thereby, the genetic differences of herring within the White Sea appear to be due, in part, to isolation during spawning time, the restriction of larvae dispersion from the spawning grounds and, potentially, because of the local adaptation to environmental habitat conditions.
Stock management
A lot of scientific attention has stressed the importance of including genetic information of marine fish in stock management. The majority of future attention needs to focus on identifying evolutionarily significant units—homogenous gene pools. Moreover, observations of lost genetic diversity and genetic breaks across multiple species should have strong implications for ecosystem management (Carvalho and Hauser 1999; McPherson et al. 2001; Laikre et al. 2005; Johannesson and André 2006; Larsson et al. 2010; Limborg et al. 2012).
Our results provide evidence for heterogeneity in the levels of genetic differentiation among C. pallasii from the White Sea, Barents Sea and Kara Sea, suggesting the existence of significant genetic population structuring within this species. This differentiation is particularly noteworthy, as the two sympatric herring populations from the White Sea, C. p. marisalbi belong to the same herring fisheries management unit and are not separated by geographical distance. If management units are intended to include only one genetically discrete group of herring, the White Sea fishery should be conducted in a manner that conserves the diversity of both spring- and summer-spawning populations. Due to this, the management units in this region warrant re-consideration.
As no population structure was observed in the Chesha–Pechora and Kara herring, there is no genetic basis for management currently being conducted. However, these herring may be vulnerable to loss of genetic variation because they should be protected through careful management. The best strategy to promote the long-term survival of C. p. suworowi is therefore to protect and sustain local stocks.
Summary
There is evidence to suggest genetic structuring and heterogeneity in levels of differentiation among C. pallasii within the northeast Atlantic area. Herring are categorised into three major groups: the sympatric spring-spawning and the summer-spawning herring, C. p. marisalbi, in the inner White Sea, and the spring-spawning herring, C. p. suworowi, in the Barents and Kara seas. We hypothesise that temporal isolation and restriction of larvae dispersion promoted reproductive isolation in the sympatric spawning groups, fostering genetic divergence. We also hypothesise that straying promoted genetic cohesion among Chesha–Pechora herring groups, and the existing physical barriers limit the gene flow between C. p. marisalbi and C. p. suworowi.
References
ACIA (2004) Impacts of a warming Arctic: Arctic climate impact assessment. ACIA overview report. Cambridge University Press, Cambridge
Altukhov KA (1958) Fishes of the White Sea (in Russian). Nauka, Moscow
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinforma 9:323
Avise JC (1994) Molecular markers, natural history and evolution. Chapman and Hall, New York
Beacham TD, Schweigert JF, MacConnachie C et al (2002) Population structure of herring (Clupea pallasi) in British Columbia determined by microsatellites, with comparisons to southeast Alaska and California. Can Sci Advis Secr, Res Doc 2002/109. http://www.pac.dfo-mpo.gc.ca/csas
Beerli P (2003) MIGRATE- a maximum likelihood program to estimate gene flow using the coalescent. http://people.scs.fsu.edu/~beerli/download.html
Berger V, Naumov AD (2001) General features. In: Berger V et al (eds) White Sea. Ecology and environment. Derzavets, St. Petersburg, pp 9–20
Buonaccorsi VP, McDowell JR, Graves JE (2001) Reconciling patterns of inter-ocean molecular variance from four classes of molecular markers in blue marlin (Makaira nigricans). Mol Ecol 10:1179–1196
Carvalho GR, Hauser L (1999) Molecular markers and the species concept: New techniques to resolve old disputes? Rev Fish Biol Fish 9:379–382
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Dmitriev NA (1946) Biology and fishery of herring in the White Sea (in Russian). Pishchepromizdat, Moscow
Durand JD, Collet A, Chow S, Guinand B, Borsa P (2005) Nuclear and mitochondrial DNA markers indicate unidirectional gene flow of Indo-Pacific to Atlantic bigeye tuna (Thunnus obesus) populations, and their admixture of southern Africa. Mar Biol 147:313–322
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. doi:10.1007/s12686-011-9548-7
Estoup A, Angers B (1998) Microsatellites and minisatellites for molecular ecology: theoretical and experimental considerations. In: Carvalho GR (ed) Advances in molecular ecology. IOS Press, Amsterdam, pp 55–86
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Evseenko SA, Mishin AV (2011) On the distribution of larvae and localization of spawning stocks of White Sea herring Clupea pallasii marisalbi. J Ichthyol 51:776–787
Evseenko SA, Andrianov DP, Mishin AV, Naumov AP (2006) Species composition and distribution of ichthyoplankton in the White Sea in July 2003. J Ichthyol 46:640–652
Excoffier L, Laval G, Schneider S (2005) ARLEQUIN (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Glover KA, Skaala O, Limborg M, Kvamme C, Torstensen E (2011) Microsatellite DNA reveals population genetic differentiation among sprat (Sprattus sprattus) sampled throughout the north-east Atlantic, including Norwegian fjords. ICES J Mar Sci 68:2145–2151
Goldstein DB, Roemer GW, Smith DA, Reich DE, Bergman A, Wayne RK (1999) The use of microsatellite variation to infer population structure and demographic history in a natural model system. Genetics 151:797–801
Gonzalez EG, Zardoya R (2007) Relative role of life-history traits and historical factors in shaping genetic population structure of sardines (Sardina pilchardus). BMC Evol Biol 7:197. doi:10.1186/1471-2148-7-197
Gonzalez EG, Beerli P, Zardoya R (2008) Genetic structuring and migration patterns of Atlantic bigeye tuna, Thunnus obesus (Lowe, 1839). BMC Evol Biol 8:252. doi:10.1186/1471-2148-8-252
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www.unil.ch/izea/softwares/fstat.html
Grant WS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89:415–426
Guo S, Thompson EA (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hardy OJ, Vekemans X (2002) SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Charbonnel N, Freville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hauser L, Carvalho GR (2008) Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish 9:333–362
Hauser L, Ward RD (1998) Population identification in pelagic fish: the limits of molecular markers. In: Carvalho GR (ed) Advances in molecular ecology. IOS Press, Amsterdam, pp 191–224
Hauser L, Adcock GJ, Smith PJ et al (2002) Loss of microsatellite diversity and low effective population size in an overexploited population of New Zealand snapper (Pagrus auratus). Proc Natl Acad Sci USA 99:11742–11747
Hedgecock D (1994) Does variance in reproductive success limit effective population size of marine organisms? In: Beaumont A (ed) Genetics and evolution of aquatic organisms. Chapman and Hall, London, pp 122–134
Hedrick PW (1986) Genetic polymorphism in heterogeneous environments: a decade later. Annu Rev Ecol Syst 17:535–566
Hewitt GM (1996) Some genetic consequences of ice ages and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hutchinson DW, Templeton AR (1999) Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53:1898–1914
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13. http://ibdws.sdsu.edu/
Johannesson K, André C (2006) Life on the margin: genetic isolation and diversity loss in a peripheral marine ecosystem, the Baltic Sea. Mol Ecol 15:2013–2029
Jørstad KE, Novikov GG, Stasenkova NJ et al (2001) Intermingling of herring stocks in the Barents Sea area. In: Balckburn FF et al (eds) Herring: expectations for a new millennium. Fairbanks, Alaska, pp 629–633
Koizumi I, Yamamoto S, Maekawa K (2006) Decomposed pairwise regression analysis of genetic and geographic distances reveals a metapopulation structure of stream-dwelling Dolly Varden charr. Mol Ecol 15:3175–3189
Kulakov MY, Pogrebov VB, Timofeyev SF, Chernova NV, Kiyko OA (2006) Ecosystems of the Barents and Kara seas, coastal segment. In: Robinson AR, Brin KH (eds) The sea ideas and observations on progress in the study of sea, V 14. Harvard University Press, Cambridge, pp 1135–1172
Laakkonen HM, Lajus DM, Strelkov P, Väinölä R (2013) Phylogeography of amphi-boreal fish: tracing the history of the Pacific herring Clupea pallasii in North-East European seas. BMC Evol Biol 13:67. doi:10.1186/1471-2148-13-67
Laikre L, Palm S, Ryman N (2005) Genetic population structure of fishes: implications for coastal zone management. Ambio 34:111–119
Lajus DL (1996) White Sea herring (Clupea pallasi marisalbi, Berg) population structure: interpopulation variation in frequency of chromosomal rearrangement. Cybium 20(3):279–294
Lajus DL (2002) Long-term discussion on the stocks of the White Sea herring: historical perspective and present state. ICES Mar Sci Symp 215:321–328
Larsson LC, Laikre L, André C, Dahlgren TG, Ryman N (2010) Temporally stable genetic structure of heavily exploited Atlantic herring (Clupea harengus) in Swedish waters. Heredity 104:40–51
Lesica P, Allendorf FW (1995) When are peripheral populations valuable for conservation? Conserv Biol 9:753–760
Lewis PO, Zaykin D (2001) Genetic data analysis: Computer program for the analysis of allelic data: version 1.0 (d 16c). http://lewis.eeb.unconn.edu/lewishome/software.html
Limborg MT, Pedersen JS, Hemmer-Hansen J, Tomkiewicz J, Bekkevold D (2009) Genetic population structure of European sprat, Sprattus sprattus: differentiation across a steep environmental gradient in a small pelagic fish. Mar Ecol Prog Ser 379:213–224
Limborg MT, Helyar SJ, de Bruyn M et al (2012) Environmental selection on transcriptome-derived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus). Mol Ecol 21:3686–3703
Liu M, Lin L, Gao T, Yanagimoto T, Sakurai Y et al (2012) What maintains the central North Pacific genetic discontinuity in Pacific herring? PLoS One 7(12):e50340. doi:10.1371/journal.pone.0050340
Manni F, Guérard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum Biol 76(2):173–190
Matishov G, Zuyev A, Golubev V et al (2004) Climatic atlas of the Arctic seas 2004: part I. Database of the Barents, Kara, Laptev, and White Seas-oceanography and marine biology NOAA Atlas NESDIS 58. U.S. Gov. Printing Office, Washington DC
McPherson AA, O’Reilly PT, McParland TL et al (2001) Isolation of nine novel tetranucleotide microsatellites in Atlantic herring (Clupea harengus). Mol Ecol Notes 1:31–32
Miller KM, Laberee K, Schulze AD, Kaukinen KH (2001) Development of microsatellite loci in Pacific herring (Clupea pallasi). Mol Ecol Notes 1:131–132
Mills LS, Allendorf FW (1996) The one-migrant-per-generation rule in conservation and management. Conserv Biol 6:1509–1518
Mishin AV, Evseenko SA, Evdokimov YuV (2008) Species composition and distribution of summer ichthyoplankton in Chupa Estuary (Kandalaksha Bay of the White Sea). J Ichthyol 48:80–816
O’Connell M, Dillon MC, Wright JM (1998) Development of primers for polymorphic microsatellite loci in the pacific herring (Clupea harengus pallasi). Mol Ecol 7:358–360
Olafsson K, Pampoulie C, Hjorleifsdottir S, Gudjonsson S, Hreggvidsson GO (2014) Present-day genetic structure of Atlantic Salmon (Salmo salar) in Icelandic rivers and Ice-Cap retreat models. PLoS One 9(2):e86809
Olsen JB, Lewis CJ, Kretschmer EJ et al (2002) Characterization of 14 tetranucleotide microsatellite loci derived from Pacific herring. Mol Ecol Notes 2:101–103
Palstra FP, Fraser DJ (2012) Effective/census population size ratio estimation: a compendium and appraisal. Ecol Evol 2(9):2357–2365
Pantyulin AN (2003) Hydrological system of the White Sea. Oceanology 43:1–14
Peery MZ, Kirby R, Reid BN et al (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Piry S, Luikart G, Conuet JM (1999) Bottleneck: a computer program for detecting recent reduction in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86(5):248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Ruzzante DE, Mariani S, Bekkevold D et al (2006) Biocomplexity in a highly migratory pelagic marine fish, Atlantic herring. Proc R Soc B 273:1459–1464. doi:10.1098/rspb.2005.3463
Ryman N, Palm S, André C, Carvalho GR, Dahlgren TG et al (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:2031–2045
Schwartz MK, Mills LS, Ortega Y, Ruggiero LF, Allendorf FW (2003) Landscape location affects genetic variation of Canada lynx (Lynx canadensis). Mol Ecol 12:1807–1816
Semenova AV, Andreeva AP, Karpov AK, Frolov SB, Feoktistov EI, Novikov GG (2004) Genetic variation of herrings of the genus Clupea from the White Sea. J Ichthyol 44:229–238
Semenova AV, Andreeva AP, Karpov AK, Novikov GG (2009) An analysis of allozyme variation in herring Clupea pallasii from the White and Barents seas. J Ichthyol 49:313–330. doi:10.1134/S0032945209040043
Semenova AV, Andreeva AP, Karpov AK, Stroganov AN, Rubtsova GA, Afanasiev KI (2013) Analysis of microsatellite loci variations in herring (Clupea pallasii marisalbi) from the White Sea. Russ J Genet 49:652–666. doi:10.1134/S1022795413060100
Semenova AV, Stroganov AN, Smirnov AA, Afanasiev KI, Rubtsova GA (2014) Genetic variation of herring Clupea pallasii from the Sea of Okhotsk revealed by microsatellite. Russ J Genet 50:65–69. doi:10.1134/S1022795413120107
Shikano T, Ramadevi J, Merilä J (2010) Identification of local- and habitat-dependent selection: scanning functionally important genes in nine-spined sticklebacks (Pungitius pungitius). Mol Biol Evol 27(12):2775–2789
Shrimpton JM, Health DD (2003) Census vs. effective population size in chinook salmon: large- and small-scale environmental perturbation effects. Mol Ecol 10:2571–2583
Small MP, Loxterman JL, Frye AE, Von Bargen JF, Bowman C, Young SF (2005) Temporal and spatial genetic structure among some Pacific herring (Clupea pallasi) populations in Puget Sound and the Southern Strait of Georgia. Trans Am Fish Soc 134:1329–1341. doi:10.1577/T05-050.1
Sugaya T, Sato M, Yokoyama E et al (2008) Population genetic structure and variability of Pacific herring Clupea pallasii in the stocking area along the Pacific coast of northern Japan. Fish Sci 74:579–588. doi:10.1111/j.1444-2906.2008.01561.x
Sunnuks P (2000) Efficient genetic markers for population biology. Trends Ecol Evol 15:199–203
Svetovidov AN (1952) Clupeidae. In: Fauna SSSR. The fishes 2(1) (In Russian). Zoologicheskii Institut Akademiya Nauk SSSR, Moscow
Teacher AGF, Kähkönen K, Merilä J (2011) Development of 61 new transcriptome-derived microsatellites for the Atlantic herring (Clupea harengus). Conserv Genet Resour. doi:10.1007/s12686-011-9477-5
Teacher AGF, André C, Jonsson PR, Merilä J (2013) Oceanographic connectivity and environmental correlates of genetic structuring in Atlantic herring in the Baltic Sea. Evol Appl. doi:10.1111/eva.12042
Vähä J-P, Erkinaro J, Niemelä E, Primmer CR (2007) Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol 16:2638–2654
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Ward RD, Woodmark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater and anadromous fishes. J Fish Biol 44:213–232
Warnock WG, Rasmussen JB, Taylor EB (2010) Genetic clustering methods reveal bull trout (Salvelinus confluentus) fine-scale population structure as a spatially nested hierarchy. Conserv Genet 11:1421–1433
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whittaker JC, Harbord RM, Boxall N et al (2003) Likelihood-based estimation of microsatellite mutation rates. Genetics 164(2):781–787
Wildes SL, Vollenweider JJ, Nguyen HT, Guyon JR (2011) Genetic variation between outer-coastal and fjord populations of Pacific herring (Clupea pallasii) in the eastern Gulf of Alaska. Fish Bull 109:382–393
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (Grant No. 13-04-00247-a) and the Leading Scientific Schools Program (Project NSh2666.2014.4).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Semenova, A.V., Stroganov, A.N., Afanasiev, K.I. et al. Population structure and variability of Pacific herring (Clupea pallasii) in the White Sea, Barents and Kara Seas revealed by microsatellite DNA analyses. Polar Biol 38, 951–965 (2015). https://doi.org/10.1007/s00300-015-1653-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-015-1653-8