
Abstract
Low-level laser therapy is commonly used to treat tendinopathy or tendon injury. Tendon healing requires tenocyte migration to the repair site, followed by proliferation and synthesis of the extracellular matrix. There are few evidence to elucidate that low-level laser promote tenocyte proliferation. This study was designed to determine the effect of laser on tenocyte proliferation. Furthermore, the association of this effect with secretion of nitric oxide (NO) and the expressions of proliferating cell nuclear antigen (PCNA) and cyclins D1, E, A, and B1 was investigated. Tenocytes intrinsic to rat Achilles tendon were treated with low-level laser (660 nm). Tenocyte proliferation was evaluated by MTT assay and immunocytochemistry with Ki-67 stain. NO in the conditioned medium was measured by ELISA. Western blot analysis was used to evaluate the protein expressions of PCNA and cyclins D1, E, A, and B1. The results revealed that tenocytes proliferation was enhanced dose dependently by laser. NO secretion was increased after laser treatment. PCNA and cyclins E, A, and B1 were upregulated by laser. In conclusion, low-level laser irradiation stimulates tenocyte proliferation in a process that is mediated by upregulation of NO, PCNA, and cyclins E, A, and B1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Low-level laser therapy (LLLT) has been used to treat musculoskeletal pain for nearly 3 decades [1]. Clinical applications show its potential of effectiveness in treating soft tissue musculoskeletal injuries, chronic pain, and wound healing [2]. Many studies have revealed the effectiveness of LLLT to treat patients with tendinopathy [3–6]. Although the clinical usage of laser suggests its efficacy, scientific evidence of its underlying molecular mechanisms for tendinopathy treatment remained limited.
In vivo studies have revealed that LLLT could enhance Achilles tendon healing by improving collagen fiber organization, preventing oxidative stress, and reducing fibrosis [7–11]. The exact mechanism is still being explored, but it is likely that the mechanism is photochemical rather than thermal [12–14]. In the regenerative phase of tendon injury, the tenocytes migrate into the repaired site, proliferate actively, and are responsible for the abundant deposition of extracellular matrix (ECM). It was demonstrated that laser irradiation could promote porcine tenocyte proliferation and upregulation of type I collagen and decorin [15]. However, to our knowledge, there are few studies exploring the molecular mechanism of laser irradiation on tenocyte proliferation.
Light irradiation can alter the redox state of cells and expression of redox-sensitive transcription factors like nuclear factor kappa B (NF-κB), which can activate inducible form of nitric oxide synthase (iNOS) and lead to a proliferation increase [16, 17]. Cell proliferation is governed by the eukaryotic cell cycle [18], which is regulated by a variety of signals that act to inhibit cell cycle progression. Tenocytes, as other cells, have four cell division cycle stages: gap 1 (G1), synthesis (S), G2, and mitosis (M). This tightly controlled temporal order is imposed by the sequential activation of a number of protein kinases known as Cdks by forming complex with various cyclins [19]. Meanwhile, proliferating cell nuclear antigen (PCNA), the auxiliary protein of DNA polymerases δ and ε, is essential for DNA replication and repair [20]. PCNA mRNA is present in all phases of the cell cycle, with exponential cell growth and a two to threefold increase during the S phase [21]. Blocking PCNA production inhibits cell division, indicating that PCNA is pivotal in the process of cell proliferation [22]. Therapeutic ultrasound can upregulate PCNA expression, thus enhance tenocytes proliferation [23]. Whether laser can stimulate tenocyte proliferation and such effect is related to expressions of nitric oxide (NO), PCNA, or cyclins have never been investigated.
The purpose of this study is to investigate the effect of laser on tenocyte proliferation and its association with the expressions of NO, PCNA, and cyclins D1, E, A, and B1.
Methods
All the procedures were approved by Chang Gung University Institutional Animal Care and Use Committee before the experiments.
Primary culture of rat Achilles tenocytes
The Achilles tendons from 16 Sprague–Dawley rats (weighing 200 to 250 g) were excised. The excised tendon was soaked in povidone-iodine for 3 min and washed twice in phosphate-buffered saline (PBS). Each tendon was then cut into small pieces of approximately 1.5–2.0 mm3 (six pieces in total). These pieces were individually placed in six-well culture plates.
After 5 min of air-drying for better adherence, 0.5 ml of Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Logan, UT, USA), with 10 % fetal bovine serum (FBS) (Cansera, Rexdale, ON, Canada), 100 U/ml penicillin, and 100 μg/ml streptomycin, was added to each well. The explants were then incubated at 37 °C in a humidified atmosphere of 5 % CO2/95 % air. After migrating out from the explants, the cells started to grow rapidly, and the confluence culture was subcultured by trypsin digestion at a 1:3 dilution ratio. Tenocytes from passages 2 and 4, with proper growth rate and normal fibroblast shape, were used. All the experiments were performed in triplicate.
Laser irradiation procedure
Laser irradiation was carried out with 660-nm laser (Konftec, Megalas-AM-800, New Taipei City, Taiwan) in continuous mode with output power of 50 mW and unit energy density of 0.0032 J/s/cm2. The power of irradiation was uniformly checked with a power meter before and after treatment. The laser beam irradiated the culture plate from above with a distance of 30 cm and covered the area up to 314 cm2. The laser beam was emitted perpendicularly and evenly to the culture plates for three groups of wells for periods of 5.2, 7.8, 10.4, and 13.0 min, respectively. The corresponding energy densities were 1.0, 1.5, 2.0, and 2.5 J/cm2. The irradiation was done on a clean bench through the culture medium at room temperature. The control groups were not subjected to laser irradiation but were removed from the incubator for the same time period as laser-treated plates.
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay
Tenocytes without or with different doses of laser treatment were cultured for 24 h. Cell proliferation was determined by MTT assay. MTT (50 μg/ml) was added and incubated at 37 °C for 1 h. Then, the MTT solution was discarded, and 1 ml DMSO was added to dissolve formazan crystals. Optical density (OD) at 570 nm (OD 570 nm) in aliquots was read using a spectrophotometer. Relative fold change of OD value for laser-treated tenocytes relative to control tenocytes was calculated.
Immunocytochemistry with Ki-67 stain
For the purpose of direct microscopic examination on the results of immunocytochemical analysis, tenocytes were subcultured and then seeded directly on the glass coverslips. Subconfluent tenocytes were then treated without and with 1.0 and 2.0-J/cm2 laser. After 24 h, all immunostaining procedures were performed as previously described [23]. Finally, tenocytes were incubated for 1 h with rabbit monoclonal antibody against Ki-67 (Nunc, Thermo-scientific, Rockford, IL, USA) diluted in blocking solution. The signal was detected with goat antirabbit IgG fluoresein (FITC)(Leinco Technologies, Inc., St. Louis, MI, USA). After washing in 1× PBS, cells were stained in 1× PBS containing 4 μg/ml propidium iodide (PI) and 100 μg/ml DNase-free RNase for 30 min at 37 °C. After washing in 1× PBS, the fluorescent PI-stained nuclei were examined under fluorescent microscope (100×). The percentage of Ki-67 positive cells was defined as the number of nuclei that stained with fluorescent green/total number of cells on the coverslip × 100 %.
Nitric oxide measurement
NO measurement was performed using NO detection kit (iNtRON Biotechnology, Seongnam, Korea), which detects NO by indirectly measuring nitrite, a byproduct of nitric oxide in the conditioned medium with or without laser treatment. This kit is based on the colorimetric change at absorbance of 490 nm, which occurs when naphthylethylenediamine is added to the byproduct of reaction between sulfanilamide and nitrite.
Western blot analysis
Cell extracts were prepared in lysis buffer containing Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2 mM DTT, 2 mM PMSF, and 1 % Triton X-100 followed by sonication method. Protein concentration of the cell extracts was determined by Bradford assay (Bio-Rad Laboratories, CA, USA). Samples with identical protein quantities were then separated by 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a PVDF membrane. The membrane was incubated at room temperature in blocking solution (1 % BSA, 1 % goat serum in PBS) for 1 h, followed by 2-h incubation in blocking solution containing an appropriate dilution of primary antibody, e.g., antitubulin, PCNA, and cyclins D1, E, A, and B1 (NeoMarks, Fremont, CA, USA). After washing, the membrane was incubated in PBS containing goat antimouse IgG conjugated with horseradish peroxidase (Sigma, St. Louis, MO, USA) for 1 h. The membranes were washed, and the positive signals developed with enhanced chemiluminescence reagent (Amershan Pharmacia Biotech, Little Chalfont Buckinghamshire, UK). The semiquantitative measurement of the band density was calculated by Digital Analysis Software (Kodak Digital Science TM, Eastman Kodak, Rochester, NY). The band density of each protein was normalized to relative band density of tubulin.
Statistical analysis
All data were expressed as mean ± SEM. Comparisons between the results of MTT assays and densitometric analysis for Western blot analysis of the laser-treated and control cells were performed using Kruskal-Wallis test. A Mann-Whitney test was used to identify where the difference occurred. The level of statistical significance is set at a p value of 0.05.
Results
Effect of laser on tenocyte proliferation
Figure 1 revealed that laser could increase the number of viable cells in a dose-dependent manner (p = 0.041). The percentage of OD value relative to the control group was 102.2 ± 2.5, 103.6 ± 3.0, 112.8 ± 3.3, and 109.6 ± 8.2 % for tenocytes treated with 1.0, 1.5, 2.0, and 2.5 J/cm2, respectively (Fig. 1). Laser at 2.0 J/cm2 could result in the most significant cell proliferation. Immonocytochemistry revealed much more tenocytes treated with laser stained positively with fluorescent green (Fig. 2a), which indicates that laser enhanced tenocyte proliferation. It also revealed that the percentage of Ki-67 positive tenocytes increased dose dependently after laser treatment (Fig. 2b). In the control group, the percentage of Ki-67 positive tenocytes were 53.6 ± 9.1, 66.4 ± 10.0, and 76.4 ± 0.7 % in control group, tenocytes treated with 1 and 2 J/cm2, respectively. The difference was statistically significant (p < 0.05).

Result of MTT assay revealed that laser irradiation increased the number of viable tenocytes in a dose-dependent manner (*p < 0.05)

a Immunocytochemistry with Ki-67 (green) and PI stain (red) in control tenocytes (left) and b tenocytes with laser treatment (2 J/cm2, right) (100×). b The percentage of Ki-67 positive tenocytes were increased dose dependently (p < 0.05)
Effect of laser on the level of secreted NO
Concentration of NO measured in the conditioned medium also revealed that laser dose dependently enhanced the level of secreted NO by tenocytes. The relative concentration of NO to control for tenocytes treated with 1.0, 1.5, 2.0, and 2.5-J/cm2 laser was 112.1 ± 10.6, 114.3 ± 6.9, 122.9 ± 5.9, and 119.6 ± 1.1 %, respectively (p = 0.05; Fig. 3).

NO in the conditioned medium of tenocytes was enhanced by laser with the most significant effect at 2.0 J/cm2 (*p < 0.05)
Effect of laser on expression of PCNA and cyclins D1, E, A, and B1
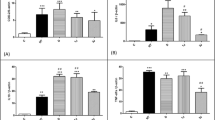
Western blot analysis and subsequent analysis revealed that laser upregulated PCNA protein expression at optimal dosage of 2.0 J/cm2. The relative protein expression of PCNA of laser-treated tenocytes to control cells was 116.6 ± 1.7, 116.7 ± 1.1, 120.4 ± 1.2, and 104.8 ± 0.9 % for tenocytes treated with 1.0, 1.5, 2.0, and 2.5-J/cm2 laser, respectively (p < 0.05; Fig. 4a, b)

a Western blot analysis revealed that PCNA was upregulated by laser irradiation. The tubulin (as internal control) and PCNA were identified at 57 and 36 kDa, respectively. b Densitometric analysis of PCNA to tubulin ratio (*p < 0.05)
Western blot analysis revealed that protein expression of cyclin E, A, and B1 was enhanced by laser irradiation (Fig. 5a). However, the expression of cyclin D was not enhanced by laser (Fig. 5a, b). Band densitometry analysis for cyclin E, A, and B1 revealed that 2.0 (113.2 ± 1.1 %), 2.5 (114.5 ± 0.8 %), and 2.0 J/cm2 (124.6 ± 13.7 %), respectively, were the most optimal doses to upregulate the most significant corresponding protein expressions (Fig. 5c, d, e; p < 0.05).

a Western blot analysis revealed that the expressions of cyclin E, cyclin A, and cyclin B1 were upregulated by laser irradiation, whereas that of cyclin D1 was not upregulated. The tubulin (as internal control) and cyclins D1, E, A, and B1 were identified at 57, 36, 47, 60, and 62 kDa, respectively. Densitometric analysis of cyclin D1, cyclin E, cyclin A, and cyclin B1 to tubulin ratio (*p < 0.05) was revealed on figures b, c, d, and e, respectively
Discussion
Tendon structure consists mainly of dense collagen arranged in a linear fashion and the basic cellular component, i.e., tenocytes (tendon cells; fibroblasts). Tenocytes, appearing as a stellate cell in cross sections and in rows in longitudinal section, are the source of collagen production, protein mediators of repair, and matrix proteoglycans [24, 25]. For an injured tendon, the healing process can be divided into three overlapping phases: (1) inflammation, (2) regeneration, and (3) remodeling and maturation. In the regenerative phase of tendon injury, tenocytes migrate into the repaired site, proliferate actively, and are responsible for the abundant deposition of ECM in the tissue. Tenocyte proliferation is fundamental to the healing process of an injured tendon. To our knowledge, this study was the first to demonstrate that laser, in a dose-dependent manner, can enhance tenocytes proliferation, which is associated with the upregulation of expressions of PCNA and cyclin E, cyclin A, and cyclin B1.
NO, a small free radical generated by nitric oxide synthases, is induced after tendon injury especially by healing fibroblasts [26]. In animal models, inhibition of NOS results in reduced tendon healing, and the addition of NO, on the other hand, enhances the healing of tendon [27]. Therefore, the local delivery of NO has been considered as a therapeutic option for tendon injury [28]. In the present study, we demonstrated that the level of secreted NO in tenocytes could be induced by the treatment with low-level laser in a dose-dependent manner. Mitochondria are thought to be the major photoreceptors for laser that trigger the initial events including increase of adenosine triphosphate, reactive oxygen species, intracellular calcium, and release of NO, which then leads to the expression of genes involved in the process of proliferation [29].
Cyclin E is induced at the G1/S boundary; Cyclin A is induced at a later phase of the cell cycle and is required for the cell to progress through the S phase [30]. Cyclin B1 regulated transition through the G2/M checkpoint of the cell cycle [31]. PCNA is identified as essential for the coordinated synthesis of leading and lagging DNA strands [20]. Besides involvement in the DNA synthesis and repair pathways, PCNA is shown to establish interactions with proteins required for the control of the cell cycle [32]. The findings of this study indicated that low-level laser might promote tenocytes to pass through G1 phase, enter S phase and transition through the G2/M checkpoint by affecting early cell cycle regulatory genes, and thus promotes cell proliferation.
The laser dose used in this study was between 1.0 and 2.5 J/cm2, which is within the recommended dose range (0.1 to 3.0 J/cm2) to treat tendinopathy.[33] The upper limit of the laser dose in our study was 2.5 J/cm2 because higher doses (>2.5 J/cm2) would decrease cell viability and damage tenocytes in this experimental setting. This finding was similar to the report that laser increases cell proliferation of Achilles tendon fibroblasts, with the optimal dose being 2.0 J/cm2. Higher doses of laser stimulation (3.0 J/cm2) do not enhance cell proliferation on Achilles tendon fibroblasts [15].
There are some limitations for this study. First, the results of this study were investigated after 24 h of laser exposure. It is uncertain as to the accumulated effects of repeated laser treatments for a longer period of time on tenocytes, which is a common practice to treat patient with tendinopathy or tendon injury. Second, it should be cautious when extrapolating these in vitro results to in vivo condition. Therefore, further animal studies are mandatory to verify the findings of this study. However, a thorough understanding of the dose effects and molecular mechanism of laser on tenocyte migration does provide laboratory-based evidence to support the use of laser to treat tendinopathy or tendon injury.
Conclusion
Laser irradiation can enhance tenocyte proliferation with increased level of secreted NO, upregulation of PCNA, as well as cyclins E, A, and B1. These findings provide novel molecular mechanisms underlying LLLT for treating tendinopathy or tendon injury.
References
Goldman JA (1981) Investigative studies of laser technology in rheumatology and immunology. In: Goldman L (ed) The biomedical laser: technology and clinical applications. Springer-Verlag, New York, p 293
Basford JR, Baxter GD (2010) Therapeutic physical agents. In: Frontera WR (ed) DeLisa’s physical medicine and rehabilitation, 5th edn. Lippincott Williams & Wilkins, Philadelphia, pp 1706–1707
Tumilty S, Munn J, McDonough S, Hurley DA, Basford JR, Baxter GD (2010) Low level laser treatment of tendinopathy: a systematic review with meta-analysis. Photomed Laser Surg 28:3–16
Bjordal JM, Lopes-Martins RA, Joensen J, Couppe C, Ljunggren AE, Stergioulas A, Johnson MI (2008) A systematic review with procedural assessments and meta-analysis of low level laser therapy in lateral elbow tendinopathy (tennis elbow). BMC Musculoskelet Disord 9:75
Stergioulas A, Stergioula M, Aarskog R, Lopes-Martins RA, Bjordal JM (2008) Effects of low-level laser therapy and eccentric exercises in the treatment of recreational athletes with chronic achilles tendinopathy. Am J Sports Med 36:881–887
Saunders L (2003) Laser versus ultrasound in the treatment of supraspinatus tendinosis: randomised controlled trial. Physiotherapy 89:365–373
Casalechi HL, Nicolau RA, Casalechi VL, Silveira L Jr, De Paula AM, Pacheco MT (2009) The effects of low-level light emitting diode on the repair process of Achilles tendon therapy in rats. Lasers Med Sci 24:659–665
Oliveira FS, Pinfildi CE, Parizoto NA, Liebano RE, Bossini PS, Garcia EB, Ferreira LM (2009) Effect of low level laser therapy (830 nm) with different therapy regimes on the process of tissue repair in partial lesion calcaneous tendon. Lasers Surg Med 41:271–276
Fillipin LI, Mauriz JL, Vedovelli K, Moreira AJ, Zettler CG, Lech O, Marroni NP, Gonzalez-Gallego J (2005) Low-level laser therapy (LLLT) prevents oxidative stress and reduces fibrosis in rat traumatized Achilles tendon. Lasers Surg Med 37:293–300
Nouruzian M, Alidoust M, Bayat M, Bayat M, Akbari M (2013) Effect of low-level laser therapy on healing of tenotomized Achilles tendon in streptozotocin-induced diabetic rats. Lasers Med Sci 28:399–405
Guerra Fda R, Vieira CP, Almeida MS, Oliveira LP, de Aro AA, Pimentel ER (2013) LLLT improves tendon healing through increase of MMP activity and collagen synthesis. Lasers Med Sci 28:1281–1288
Karu T (1999) Primary and secondary mechanisms of action of visible to near-IR radiation on cells. J Photochem Photobiol B 49:1–17
Lane N (2006) Cell biology: power games. Nature 443:901–903
Tafur J, Mills PJ (2008) Low-intensity light therapy: exploring the role of redox mechanisms. Photomed Laser Surg 26:323–328
Chen CH, Tsai JL, Wang YH, Lee CL, Chen JK, Huang MH (2009) Low-level laser irradiation promotes cell proliferation and mRNA expression of type I collagen and decorin in porcine Achilles tendon fibroblasts in vitro. J Orthop Res 27:646–650
Alexandratou E, Yova D, Handris P, Kletsas D, Loukas S (2002) Human fibroblast alterations induced by low power laser irradiation at the single cell level using confocal microscopy. Photochem Photobiol Sci 1:547–552
Lubart R, Breitbart H (2000) Biostimulative effects of low-energy lasers and their implications for medicine. Drug Dev Res 50:471–475
Sherr CJ (1996) Cancer cell cycles. Science 274:1672–1677
Pines J (1994) Protein kinases and cell cycle control. Semin Cell Biol 5:399–408
Prelich G, Stillman B (1988) Coordinated leading and lagging strand synthesis during SV40 DNA replication in vitro requires PCNA. Cell 53:117–126
Morris GF, Mathews MB (1989) Regulation of proliferating cell nuclear antigen during the cell cycle. J Biol Chem 264:13856–13864
Sachs F (1991) Mechanical transduction by membrane ion channels: a mini review. Mol Cell Biochem 104:57–60
Tsai WC, Hsu CC, Tang FT, Chou SW, Chen YJ, Pang JH (2005) Ultrasound stimulation of tendon cell proliferation and upregulation of proliferating cell nuclear antigen. J Orthop Res 23:970–976
O’Brien M (1992) Functional anatomy and physiology of tendons. Clin Sports Med 11:505–520
Leadbetter WB (1992) Cell-matrix response in tendon injury. Clin Sports Med 11:533–578
Murrell GA, Szabo C, Hannafin JA, Jang D, Dolan MM, Deng XH, Murrell DF, Warren RF (1997) Modulation of tendon healing by nitric oxide. Inflamm Res 46:19–27
Bokhari AR, Murrell GA (2012) The role of nitric oxide in tendon healing. J Shoulder Elbow Surg 21:238–244
Nichols SP, Storm WL, Koh A, Schoenfisch MH (2012) Local delivery of nitric oxide: targeted delivery of therapeutics to bone and connective tissues. Adv Drug Deliv Rev 64:1177–1188
Hashmi JT, Huang YY, Osmani BZ, Sharma SK, Naeser MA, Hamblin MR (2010) Role of low-level laser therapy in neurorehabilitation. Phys Med Rehabil 2:S292–S305
Girard F, Strausfeld U, Fernandez A, Lamb NJ (1991) Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 67:1169–1179
Minshull J, Pines J, Golsteyn R, Standart N, Mackie S, Colman A, Blow J, Ruderman JV, Wu M, Hunt T (1989) The role of cyclin synthesis, modification and destruction in the control of cell division. J Cell Sci Suppl 12:77–97
Xiong Y, Zhang H, Beach D (1992) D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell 71:505–514
Bjordal JM, Couppe C, Ljunggren AE (2001) Low level laser therapy for tendinopathy: evidence of a dose response. Phys Ther Rev 6:91–99
Acknowledgments
We would like to thank the National Science of Council, Taiwan, for the financial support of this research (grant no. 99-2314-B-182A-108).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsai, WC., Cheng, JW., Chen, JL. et al. Low-level laser irradiation stimulates tenocyte proliferation in association with increased NO synthesis and upregulation of PCNA and cyclins. Lasers Med Sci 29, 1377–1384 (2014). https://doi.org/10.1007/s10103-014-1528-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-014-1528-1