
Abstract
The worldwide rise in antibiotic resistance necessitates the development of novel antimicrobial strategies. This study aimed to evaluate the bactericidal action of an 810-nm diode laser in a cutaneous wound infection. An Escherichia coli strain was transformed with a shuttle vector (pRB474) containing firefly luciferase gene from Photinus pyralis resulting in a bioluminescent phenotype. Because firefly luciferase is an enzyme and as such is prone to inactivation at elevated temperature, the first phase has consisted in evaluating in vitro the effect of temperature elevation (30, 40, 50, and 60°C for 2 min) on bacteria bioluminescence. The second phase was performed in vivo. Two full-thickness circular, 14-mm diameter wounds (control and laser-irradiated) were induced on rats. Wound infection was carried out using a suspension (50 μl PBS) containing 5x107 cells of bioluminescent E. coli (109 cells/ml). Thirty minutes later, light irradiation was performed with an 810-nm diode laser (P=10 W, ∅=1.4 cm, fluence: 130, 195, and 260 J/cm2). Temperature was measured within each wound with a noncontact infrared thermometer. Light emission of the bioluminescent bacteria was monitored in vivo by a bioluminescence imaging system before and at 4, 8, 24, and 48 h after laser irradiation. In vitro, bacteria bioluminescence is not affected when temperature is maintained at 50°C for 2 min. In vivo, bioluminescence imaging showed that at 4 h, the viability of E. coli was reduced when compared to the control (CTRL) group (p<0.01). This observation was confirmed at 8 h (p<0.001), at 24 h (p<0.001), and finally at 48 h (p<0.001). Loss of viability of E. coli depends on laser fluence. At 48 h, bioluminescent bacteria were not detected (100% loss of viability) in the wound irradiated at 260 J/cm2. For this fluence, the temperature reached 45°C at the end of the irradiation. This study confirms previous observations on the bactericidal effect of diode lasers. Because a progressive desiccation of the superficial dermis is usually observed when using laser irradiation, the hypothesis that laser irradiation dries out the wound making the wound an inhospitable place for bacteria is much more relevant than a direct effect of infrared light on chromophores inside bacteria. This is confirmed by the fact that in this latter case, one would expect an immediate drop in luminescence followed by an increase as the surviving bacteria started to divide and repopulate the wound. However, the exact mechanism deserves further studies. This study points out the advantage of using bioluminescence imaging to evaluate laser for the treatment of acute infections in vivo, nondestructively, and noninvasively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laser irradiation, apart from conventional methods, has been shown to have potential to eliminate bacteria. The antimicrobial effect of the Nd:YAG laser has been demonstrated in several in vitro studies. Schultz et al. [1], with a high power (120 W) Nd:YAG laser, reduced the viable counts of bacteria in aqueous suspension in microtiter plates. Using the same wavelength, Grönqvist et al. [2] have shown Staphylococcus epidermidis growth inhibition on agar plates. Ward et al. [3] observed a bactericidal action of high power Nd:YAG laser light on transparent and turbid Escherichia coli bacterial suspensions. Similarly, Lee and Pinheiro have used a CO2 laser to treat bacteria-infected cutaneous wounds [4].
Optical techniques have recently been proposed for rapid monitoring of the effectiveness of therapeutic strategies used in wound infection. The method of optically monitoring bacterial numbers and viability, in real time, in living animals by use of genetically engineered bacteria that emit luminescence, together with ultrasensitive photon-counting cameras, has been demonstrated in several models [5–9]. Quantification of the luminescence images can determine, in real time, the extent of infection in living animals and thereby can provide both temporal and spatial information about the labeled bacteria and their metabolic activities. Using this technique, we have recently demonstrated that antibiotic effects can be detected directly, nondestructively, and noninvasively in vivo in a cutaneous wound model [10].
This study aimed to evaluate the bactericidal action of an 810-nm diode laser in a cutaneous wound infection using a bioluminescence imaging technique. Because firefly luciferase is an enzyme and as such is prone to inactivation at elevated temperature, in vitro studies were performed first to evaluate the potential role of heat on bacteria bioluminescence.
Materials and methods
Bacterial strain
A relatively nonpathogenic strain of E. coli, which lacks virulence factors necessary to cause invasive infection, was used [11]. E. coli TOP10F′, a facultative anaerobe (ref C615-00) in the normal intestinal flora of humans and animals [12], was obtained from Invitrogen (Invitrogen SARL, Cergy Pontoise, France) and routinely grown at 37°C. Furthermore, TOP10F′ is a recombination negative strain designed for stable replication of high copy number plasmids [13]. Bioluminescent E. coli was generated by transforming the strain TOP10F′ with a firefly luciferase gene (Promega (E1781) using the shuttle vector pRB474 [14]. pRB474 was introduced into the cells by electroporation as previously described [10]. Transformants were plated onto agar plates containing 50 μg /ml of ampicillin and grown for 1 day at 35°C. Bioluminescent colonies were selected using a luminometer (Lumat LB 9501 Berthold, Berthold France SA, Thoiry, France).
In vitro studies
One hundred microliters of fresh Luria Broth containing the bioluminescent bacteria in growth phase (OD600: 0.6) was placed in a cuvette and was incubated for 2 min at 30°C (n=4), 40°C (n=4), 50°C (n=4), and 60°C (n=4) using a thermostated circulating bath (Ministat, Huber, Rimsting, Germany). The temperature was controlled inside the cuvette using a 0.2-mm thermocouple probe (Mini-Hypodermic Thermocouple Probe Model HYP-0, Omega, Guyancourt, France). The aliquots were transferred to a 96-well black sides plate for bioluminescence imaging up to 100 h postincubation. The temperature of the bioluminescence imaging chamber was maintained at 30°C. No heated aliquots (n=4 maintained at 20°C) were used as control. Before each measurement, 10 μl of substrate solution (1 mM of d-luciferin to 0.1 M phosphate–citrate buffer) was added to the well. Serial dilution and plating were used to determine the number of live bioluminescent bacteria present in the sample after heating at different temperatures at 4 and 8 h. Counts produced after plating of serially diluted product were reported as “colony-forming units” (cfu) as described by Jett [15].
Animals
All animal experiments conformed to the Ministère de l’Agriculture et de la Forêt Resolution on the use of animals in research and were approved by the Subcommittee on Research Animal Care of the Lille Medical University (protocol 2003-35).
Male Sprague–Dawley rats (Charles River France, Les Oncins, France), weighing between 200 and 300 g, were used for this study. The back of the rat was shaved, and a depilatory cream was applied to remove any remaining hair. Two full-thickness circular, 14-mm diameter wounds were created using surgical scissors on the back of the rats while they were under general anesthesia (140 mg of ketamine per kilogram of body weight and 3 mg of chlorpromazine per kilogram). The positions of these wounds were 4 and 9 cm caudal, respectively, to the ears and placed on the midline. A Teflon chamber similar to the chamber developed by Balazs was applied around each wound and was glued into the edge of the skin with Histoacryl (B. Braun Surgical GmbH, Melsungen, Germany) and sutured (Ethicon 4-0 sutures) [16]. A sterile glass window (GF-C Whatman) was placed inside the chamber to protect the wound against other infections.
Bioluminescence imaging system
Light emission of the bioluminescent bacteria was detected in vivo by a bioluminescence imaging system (IVIS 50, Xenogen, Alameda, USA). The animal was placed inside a light tight chamber on a heated moveable platform. The field of view was adjusted to measure simultaneously the control and the laser-irradiated wounds.
Two images were taken on each animal. The first image was a 0.2-s exposure of the animal illuminated by lights located in the top of the imaging chamber. This image was referred to as a “photographic image” and was displayed as a grayscale image. The second image was a 15-s exposure of the animal taken in darkness to record low-level luminescent emission. This image was referred to as a “luminescent image” and was displayed as a pseudocolor image overlaid on the photographic image. This procedure and the acquisition parameters were kept constant for all measurements. A color bar shows the relationship between the pseudocolors of the luminescent image and the numerical values of the image data. The numerical value is proportional to the number of photons detected in each pixel. It is referred to as relative luminescence units (RLU).
Regions of interest (ROIs) were used to quantify the amount of light emission detected within each wound. The ROIs encompassed the entire surface area of each wound. In each image, two equivalent ROIs (1.5 cm2) were created to quantify the amount of light emission. Each wound was imaged at 0, 4, 8, 24, and 48 h postinfection using the bioluminescent imaging system. Before each measurement, 50 μl of substrate solution (1 mM of d-luciferin to 0.1 M phosphate–citrate buffer) was added exogenously to the wound. The difference between CTRL wounds and laser-irradiated wounds was evaluated using the Student’s t test.
Laser
The laser irradiation was performed with an 810-nm diode laser (OPC-B015-FCTS, Opto Power, Tucson, AZ, USA) coupled into a 600-μm fiber to produce a 14-mm (1/e 2) circular spot on tissue. The diameter and homogeneity of the spot was controlled using thermal paper (Linargraph direct print, Kodak, Rochester, USA). The laser power was measured with a power meter (Ophir Optronics, Jerusalem, Israel).
Before the present study, several radiant powers (W/cm2) and fluences (J/cm2) were evaluated on the wound without bacterial infection since it had been demonstrated that the application of the 810-nm diode laser may cause damage to collateral tissues when radiant power is too high [17]. Only radiant powers below 8 W/cm2 and fluences below 400 J/cm2 did not induce cell damage in a cutaneous wound (data not shown). For this reason, the maximum dose used in this study was restricted to 260 J/cm2. Irradiation was performed using a power of 10 W (radiant power: 6.5 W/cm2) and irradiation times of 20, 30, and 40 s, which produced fluences of 130, 195, and 260 J/cm2, respectively.
Infection model
Three groups of three rats were used (18 wounds). A suspension (50 μl PBS) containing 5x107 cells of mid-log phase bioluminescent E. coli (109 cells/ml) was inoculated into each wound. Thirty minutes postinoculation (duration required by the bacteria to attach to the tissue) and immediately before imaging, 50 μl of substrate solution (1 mM of d-luciferin to 0.1 M phosphate–citrate buffer) was added to the wound.
Bacterial loading was controlled using the imaging system to confirm that it was equivalent in each wound. On each rat, one wound was not irradiated (CTRL) (n=9). After 30 min, nine wounds (one of each rat) were irradiated: 20 s (n=3), 30 s (n=3), and 40 s (n=3).
Wound temperature changes after exposure to light
Because heat is thought to be a key factor in bacterial killing, temperature at the surface of the wound was measured with a noncontact infrared thermometer (KT17, Heimann GmbH, Wiesbaden, Germany) before, during, and after laser irradiation.
Results
The bioluminescence signal of E. coli measured by the luminometer was linearly proportional to bacterial cfu (as determined by serial dilution and plating) from 103 to 107 organisms (data not shown).
In vitro studies
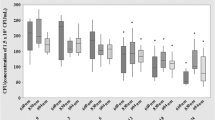
Figure 1 shows the bacteria viability measured by the bioluminescence imaging system (from 0 to 100 h). Figure 2 shows the bacteria viability measured by cfu 4 and 8 h after heating with a thermostated circulating bath for 2 min at 30°C (n=4), 40°C (n=4), 50°C (n=4), and 60°C (n=4).

Effect of the heating by thermostated bath on the viability of E. coli suspensions containing approximately 105 cfu/ml in Luria Broth. Bacterial samples were heated at different temperatures (30, 40, 50, and 60°C) during 2 min, and the loss of viability was determined by bioluminescence imaging system. Bioluminescence was recorded for 100 h

Effect of the heating by thermostated bath on the viability of E. coli suspensions containing approximately 105 cfu/ml in Luria Broth. Bacterial samples were heated at different temperatures (30, 40, 50, and 60°C) during 2 min, and the loss of viability was by colony-forming unit assay at 4 and 8 h
When compared to control, bacteria bioluminescence is not affected after heating at 30, 40, and 50°C for 2 min. Because the bacteria are in growth phase, they are able to multiply in culture medium for 25 h where a maximum bioluminescence emission is reached. Then, a continuous decline is observed due to the consumption of the culture medium.
An important reduction of bioluminescence emission from bacteria is noted in the samples heated at 60°C. However, this temperature is not sufficient to destroy all bacteria in the culture medium. This observation is confirmed by standard plate count method (Fig. 2). Figure 3 displays the bioluminescence image of the 96-well black sides plate recorded 20 h after heating.

One hundred microliters aliquots of bioluminescent bacterial suspensions recorded 20 h after heating at 30°C (n=4), 40°C (n=4), 50°C (n=4), and 60°C (n=4). Four aliquots were not heated. Control CTRL
Animal studies
Figure 4 displays the bioluminescence signal as a function of fluences and delay after irradiation. CTRL group shows a slight decrease of the bioluminescence signal as a function of time. For the three laser groups, bioluminescence imaging showed that at 4 h, the viability of bioluminescent E. coli was reduced when compared to the CTRL group (p<0.01). This observation was confirmed at 8 h (p<0.001), at 24 h (p<0.001), and finally at 48 h (p<0.001). Loss of E. coli viability depended on laser fluence. At 48 h, bioluminescent bacteria were not detected (100% loss of viability) in the wound irradiated at 260 J/cm2.

In vivo monitoring of E. coli bioluminescence in the cutaneous wound. Each wound was inoculated with a suspension (50 μl PBS) containing 5x107 cells of mid-log phase bioluminescent E. coli (109 cells/ml). Before each measurement, 50 μl of substrate solution (1 mM of d-luciferin to 0.1 M phosphate–citrate buffer) was added exogenously to the wound. Each set of data represents the mean (±SD) number of relative light units (RLU) (for each laser group: n=3, for CTRL group: n=9)
Figure 5 shows that temperature elevation was linearly proportional to the fluence used. After irradiation, surface temperatures reached 38±2°C for 130 J/cm2, 40±2.5°C for 195 J/cm2, and 45±2°C for 200 J/cm2. The regression curve has the following equation:

Wound temperature as a function of light exposure (J/cm2). The wounds infected with bioluminescent E. coli were illuminated with three different fluences: 130, 195, and 260 J/cm2. (λ=810 nm, P=10 W, ∅=14 mm)
Figure 6a,b shows two images recorded, respectively, before laser irradiation and 48 h later (CTRL and laser: 260/cm2). At 48 h, the laser-irradiated wound did not show any viable bacteria.

Overlay images recorded a before laser irradiation (above) and b 48 h later (below). Each wound was inoculated at T=0 with a suspension (50 μl PBS) containing 5x107 cells of mid-log phase bioluminescent E. coli (109 cells/ml). Before each measurement, 50 μl of substrate solution (1 mM of d-luciferin to 0.1 M phosphate–citrate buffer) was added exogenously to the wound
Discussions
In the present work, we have examined the effects of an 810-nm diode laser irradiation on a cutaneous wound infected with bioluminescent E. coli (pRB474). The firefly luciferase gene from Photinus pyralis used in this study is a single polypeptide. The light production is started by the addition of the substrate, d-luciferin. Therefore, the metabolic stress caused by light production (and ATP consumption) takes place only at the luminescence measurement stage. Recently, other bioluminescent reporter systems such as bacterial luciferase operon luxCDABE have also been used to provide a means of detecting bacterial viability [5–7, 18]. The bacteria containing operon luxCDABE produce light continuously and are under constant metabolic stress caused by light emission and/or production of five polypeptides. These reasons may explain the difference in the light production power of the firefly luciferase and the bacterial luciferase operon [19].
Using the same animal model, we have previously demonstrated that antimicrobial activity of sulfamethoxazole–trimethoprim was associated with similar changes in bioluminescence. Similarly, the slight decrease of the bioluminescence signal as a function of time observed in the CTRL group was in accordance with this previous study [10].
The in vitro study shows that the light emission of the bioluminescent bacteria was not affected by temperature increase up to 50°C maintained for 2 min. This observation was confirmed by standard plate count method. Because the bacteria were in growth phase, they were able to multiply in culture medium up to 25 h. Then, a continuous decline was observed due to the consumption of the culture medium. In addition, the bioluminescent bacteria maintained at 30°C entered a stationary growth phase more quickly than control bacteria incubated at 20°C [20, 21] A factor influencing the effectiveness of a heat treatment is the composition of the environment surrounding the bacteria. Dry and/or acidic environments increase the rate of killing at a given temperature due to the damaging effects acid has on the cell. Conversely, proteins in a solution such as medium culture have a protective effect. The D-value, which denotes the decimal reduction time, and which is considered to be the time required at a specific temperature and under specified conditions to reduce a microbial population by one decimal, has been quoted as 10 min at 54°C for E. coli [3, 22]. Consequently, this in vitro study is in agreement with previous observations. These observations confirm that temperature lower than 50°C and lasting for 2 min or less does not inactivate firefly luciferase, and thus reduction of bacteria bioluminescence must be due to bacteria inactivation or to an inappropriate medium affecting the bacterial ability to reproduce.
The in vivo study shows that bacterial killing depends on laser fluence. At 260 J/cm2, the bioluminescent bacteria were not detected in the wound after 48 h postirradiation.
It is difficult to compare the results of our study with previous laser studies because of differences in the laboratory settings and irradiation constants. Because wavelength, spot diameter, exposure time, and operating mode (CW or pulsed) are different, it is impossible to compare the lethal effect of the laser only considering the total delivered energy.
Kreisler et al. [17] have examined the cellular effects of the diode laser with a wavelength of 810 nm on human periodontal tissues. They observed that the power range (0.5–2.5 W) was not decisive for survival of cells, but rather the time of exposure. Schoop et al. [23] have evaluated various laser systems, namely, the Nd:YAG, the diode, the Er:YAG, and the Er:YSGG laser. They have demonstrated that for all the wavelengths investigated, using radiant powers greater than 1.5 W was capable of significant reductions of E. coli.
It is usually considered that the bactericidal action of the laser is due to thermal heating. Grönqvist [2], when using a Nd:YAG laser at 1,000 J/cm2, has obtained agar temperature of approximately 70°C. Because it is known that temperatures above 60°C can cause thermal damage and killing of bacteria, killing at an exposure of 1,000 J/cm2 is likely explained by photothermal mechanisms.
The D-value of E. coli at 54°C has been quoted as 10 min [22]. However, in our experiments, the maximum surface temperature reached after laser irradiation was only 45°C and was maintained for less than 1 min. Therefore, the specific question to be answered here was whether the microbial killing was simply due to heating to a lethal temperature or whether other mechanisms might contribute. Two hypotheses can be formulated:
-
(1)
Laser irradiation could induce local temperature increase inside the bacteria higher than the temperature increase measured at the surface of the wound. It has been demonstrated that chromophores inside bacteria are sensitive to infrared light. Consequently, local heating inside bacteria or light-induced modulation of enzymatic activity could be responsible for bacterial killing [24, 25]. Ward et al. also observed extensive killing with the Nd:YAG laser [3]. Temperature recording has shown that the maximum temperature was 50°C after a 23-s exposure time. Scanning electron microscopy was performed and showed that the E. coli cell surface had been injured by the laser exposure at 50°C, whereas conventional heating did not have the same effect. However, if this hypothesis were valid, one would expect an immediate drop in luminescence followed by an increase as the surviving bacteria started to divide and repopulate the wound.
-
(2)
A progressive desiccation of the superficial dermis is usually observed when using laser irradiation [26]. A dry wound surface would make the wound an inhospitable place for bacteria [27] and might explain the bacterial killing. This hypothesis seems to be more relevant because the decrease in bioluminescence is similar to the decrease observed in vitro after 25 h.
However, this later mechanism deserves further investigation. An assessment of laser-induced tissue damage should be completed with an investigation of the actual effect on the bacterial structure.
Conclusion
This study demonstrates that bioluminescent E. coli could serve as a biosensor of antibacterial activity for in vivo studies. The use of bioluminescent imaging strategies to reveal the real-time effects of potential therapeutic agents or devices on bacterial infections in cutaneous wound animal models would greatly accelerate the analyses of compounds under development. The use of diode laser irradiation on a cutaneous wound infected with bioluminescent E. coli in living animal could be proposed as an alternative antimicrobial strategy. This technique could circumvent the problems associated with the use of antibiotics such as development of resistance in target organism, including the emergence of bacterial strains that are resistant to all available antibacterial agents or permitting the colonization of opportunistic pathogens.
References
Schultz RJ, Harvey GP, Fernandez-Beros ME, Krishnamurthy S, Rodriguez JE, Cabello F (1986) Bactericidal effects of the neodymium:YAG laser: in vitro study. Lasers Surg Med 6(5):445–448
Grönqvist AWJ, Axner O, Monsen TJ (2000) Bactericidal effect of pulsed 1,064 nm Nd:YAG laser light on Staphylococcus epidermidis is of photothermal origin: an in vitro study. Lasers Surg Med (4):336–340
Ward GD, Watson IA, Stewart-Tull DE, Wardlaw AC, Wang RK, Nutley MA et al (2000) Bactericidal action of high-power Nd:YAG laser light on Escherichia coli in saline suspension. J Appl Microbiol 89(3):517–525
Lee JS, Tarpley SK, Miller AS 3rd (1999) CO2 laser sterilization in the surgical treatment of infected median sternotomy wounds. South Med J 92(4):380–384
Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA (1995) Photonic detection of bacterial pathogens in living hosts. Mol Microbiol 18(4):593–603
Francis KP, Joh D, Bellinger-Kawahara C, Hawkinson MJ, Purchio TF, Contag PR (2000) Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect Immun 68(6):3594–3600
Rocchetta HL, Boylan CJ, Foley JW, Iversen PW, LeTourneau DL, McMillian CL et al (2001) Validation of a noninvasive, real-time imaging technology using bioluminescent Escherichia coli in the neutropenic mouse thigh model of infection. Antimicrob Agents Chemother 45(1):129–137
Hamblin MR, O’Donnell DA, Murthy N, Contag CH, Hasan T (2002) Rapid control of wound infections by targeted photodynamic therapy monitored by in vivo bioluminescence imaging. Photochem Photobiol 75(1):51–57
Kadurugamuwa JL, Sin L, Albert E, Yu J, Francis K, DeBoer M et al (2003) Direct continuous method for monitoring biofilm infection in a mouse model. Infect Immun 71(2):882–890
Jawhara S, Mordon S (2004) In vivo imaging of bioluminescent Escherichia coli in a cutaneous wound infection model for evaluation of an antibiotic therapy. Antimicrob Agents Chemother 48(9):3436–3441
Theilman NM, Guerrant RL (1999) Escherichia coli. In: Yu VL, Merigan TC Jr, Barriere SL (ed) Antimicrobial therapy and vaccines. Williams & Wilkins, Baltimore, MD, pp 188–200
Bonten M, Stobberingh E, Philips J, Houben A (1990) A high prevalence of antibiotic resistant Escherichia coli in faecal samples of students in the south-east of The Netherlands. J Antimicrob Chemother 26(4):585–592
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580
Bruckner R (1992) A series of shuttle vectors for Bacillus subtilis and Escherichia coli. Gene 122(1):187–192
Jett BD, Hatter KL, Huycke MM, Gilmore MS (1997) Simplified agar plate method for quantifying viable bacteria. Biotechniques 23(4):648–650
Balazs L, Okolicany J, Ferrebee M, Tolley B, Tigyi G (2001) Topical application of the phospholipid growth factor lysophosphatidic acid promotes wound healing in vivo. Am J Physiol Regul Integr Comp Physiol 280(2):R466–R472
Kreisler M, Daublander M, Willershausen-Zonnchen B, d’Hoedt B (2001) Effect of diode laser irradiation on the survival rate of gingival fibroblast cell cultures. Lasers Surg Med 28(5):445–450
Contag CH, Spilman SD, Contag PR, Oshiro M, Eames B, Dennery P et al (1997) Visualizing gene expression in living mammals using a bioluminescent reporter. Photochem Photobiol 66(4):523–531
Hakkila K, Maksimow M, Karp M, Virta M (2002) Reporter genes lucFF, luxCDABE, gfp, and dsred have different characteristics in whole-cell bacterial sensors. Anal Biochem 301(2):235–242
Waterfield NR, Le Page RW, Wilson PW, Wells JM (1995) The isolation of lactococcal promoters and their use in investigating bacterial luciferase synthesis in Lactococcus lactis. Gene 165(1):9–15
Marincs F (2000) On-line monitoring of growth of Escherichia coli in batch cultures by bioluminescence. Appl Microbiol Biotechnol 53(5):536–541
Ingraham JL (1987) Effect of temperature, pH, water activity and pressure on growth. In: Neidhardt FC (ed) Escherichia coli and Salmonella typhimurium. American Society for Microbiology, Washington, USA, pp 1543–1554
Schoop U, Kluger W, Moritz A, Nedjelik N, Georgopoulos A, Sperr W (2004) Bactericidal effect of different laser systems in the deep layers of dentin. Lasers Surg Med 35(2):111–116
Hellingwerf KJ, Hoff WD, Crielaard W (1996) Photobiology of microorganisms: how photosensors catch a photon to initialize signalling. Mol Microbiol 21(4):683–693
Esteban B, Carrascal M, Abian J, Lamparter T (2005) Light-induced conformational changes of cyanobacterial phytochrome Cph1 probed by limited proteolysis and autophosphorylation. Biochemistry 44(2):450–461
Burkhardt BR, Maw R (1997) Are more passes better? Safety versus efficacy with the pulsed CO2 laser. Plast Reconstr Surg 100(6):1531–1534
Mertz PM, Ovington LG (1993) Wound healing microbiology. Dermatol Clin 11(4):739–747
Acknowledgements
The authors wish to thank Dr. Reinhold Brückner (Mikrobielle Genetik, Universität Tübingen, 72076 Tübingen, Germany) for kindly providing the plasmid (pRB474) and Guy Dhelin for the excellent technical assistance. The printing costs were covered by Xenogen, Alameda, USA, and the authors are particularly grateful to David Panzarella and Béatrice David.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jawhara, S., Mordon, S. Monitoring of bactericidal action of laser by in vivo imaging of bioluminescent E. coli in a cutaneous wound infection. Lasers Med Sci 21, 153–159 (2006). https://doi.org/10.1007/s10103-006-0388-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10103-006-0388-8