
Abstract
A high-performance reduced graphene oxide–polyaniline (RGO-PANI) electrode material was prepared through commercially viable, facile one-pot synthesis, applying famed diazotization chemistry and grafting strategy. In this work, 4-nitroaniline was utilized as a grafting substrate to bind reduced graphene oxide and polyaniline covalently to a highly stable and efficient supercapacitor electrode material. The chemical composition and structural analysis of covalently functionalized RGO-PANI nanocomposites were characterized by X-ray diffraction, field-emission scanning electron microscopy, Raman spectroscopy, UV–visible spectroscopy, and Fourier-transform infrared spectroscopy. The electrochemical behavior of the nanocomposites was analyzed through cyclic voltammetry, galvanostatic charge and discharge, and electrochemical impedance spectroscopy. The prepared nanocomposite shows a high specific capacitance of 490 F g−1 in 1 M Na2SO4 with outstanding cyclic stability (10,000 cycles). The applied covalent functionalization through grafting strategy was the principal factor for both high specific capacitance and excellent cyclic stability.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The utilization of fossil fuels in various machinery-based applications and its requirements has led to a worldwide energy crisis in recent decades. Additionally, humans face severe pollution problems due to unlimited consumption of fossil fuels and their harmful effluents, which is deleterious to both living organisms and the environment (Behera and Dash 2017). Nature and environment must be protected from carbon dioxide, carbon monoxide, and various greenhouse gases originating from fossil fuels. In other words, solar, wind, and tidal sources are the best alternative renewable energy resources for generating energy, reducing the utilization of non-renewable sources like coal, crude oil, and natural gas (hydrocarbons). Though energy conversion from natural resources is seasonal and limited, their good factors are no doubt preferable to radioactive nuclear power plants (Takahashi et al. 2017). Energy storage and conversion have been developing research fields in this century, and have become pivotal since the Fukushima disaster in Japan (Yamamura et al. 2017). Thus, clean energy generation and storage have become important needs in modern society. Supercapacitors are the electrochemical energy storage devices that have the potential to store and rapidly and safely deliver electric energy, with long life cycles. Recently, developing high-energy supercapacitors without sacrificing their inherent properties, including high power density, long cyclic stability, and unique charge storability, have become an important aim, due to their crucial role in energy storage and conversion, as compared to batteries and fuel cells (Wang et al. 2012a; Zhong et al. 2015). The enhancement of energy density in supercapacitors depends on either increasing cell voltage by optimizing electrolytes, or specific capacitance by electrode materials in supercapacitors (Zhang et al. 2011). In recent years, most research has focused on developing a variety of nanomaterials to build high-energy density supercapacitors, instead of exploring electrolytes (Aricò et al. 2005; Dong et al. 2016; González et al. 2016; Guo et al. 2017; Wang et al. 2012a). However, trending hybrid (or) asymmetric supercapacitors have satisfied all expected performances, including the energy and power density, launching a new electronic era. Among nanomaterials, carbon-based electrode materials have been investigated for supercapacitor applications (Wang et al. 2012b). Owing to their groundbreaking properties of high electronic conductivity, thermal conductivity, high charge carrier mobility, transparency, and mechanical strength, graphene nanocomposites have recently attracted worldwide attention (Borenstein et al. 2017; Chen et al. 2016, 2017b; Salunkhe et al. 2016; Yu et al. 2016). Many graphene- and graphene-oxide-based composites (Eng et al. 2016; Ma et al. 2016; Sumboja et al. 2013; Sun et al. 2014; Palanisamy et al. 2012, 2017a, b) along with metal chalcogenides (Xia et al. 2016), bimetallic complexes (Li et al. 2017), metal oxides (Yuan et al. 2014), metal sulfides (Xia et al. 2014), metal organic frameworks (Salunkhe et al. 2014), perovskites (Arjun et al. 2017), and conducting polymers (Basnayaka and Ram 2017) have been utilized as electrode materials in supercapacitors and sensing probes in electrochemical biosensors. The conducting polymer–graphene-based composites have been deeply exploited in supercapacitors. Remarkably, polyaniline is a well-known conducting polymer, widely used among polypyrrole, polyacetylene, poly(3,4-ethylene dioxythiophene), and polythiophene because of its high conductivity, simple preparation protocol, reliability, and inexpensive monomer (Baker et al. 2017). Polyaniline-grafted reduced graphene oxide was efficiently achieved first time by Kumar (2012) via acyl chemistry through the grafting of amine-protected 4-aminophenol on reduced graphene oxide, followed by oxidative polymerization. Then, Kumar et al. (2013) described the covalent functionalization of the graphene–polyaniline composite by utilizing 4-aminobenzoic acid as a grafting substrate to bind the graphene–polyaniline in their prolonged method. Furthermore, the same grafting strategy was adopted by Gao et al. (2014), for graphene–polyaniline, successfully changing the grafting agent P-phenylenediamine instead of 4-aminobenzoic acid. The above literature reveals the paramount importance of grafting of RGO-PANI. Apart from that, both the RGO and PANI materials are carbonaceous and degradable, without affecting the environment. From a clean energy perceptive, carbonaceous materials are preferable compared to bimetallic complexes, metal oxides, and metal sulfides. Even though various reports are available on graphene–polyaniline covalent functionalization, there is no literature regarding the simple preparation of RGO-PANI. It has inspired us to launch research on RGO-PANI. In a one-pot synthesis, we can synthesize the products in kilograms scale with less effort. From the manufacturing point of view, through a one-pot synthesis, we can scale up the designed products from kilograms to tons without changing the initial apparatus setups, with reduced human effort, electricity, and time.
In this study, the preparation contains three individual reactions, namely diazotization, reduction, and polymerization. All have performed sequentially in acetic media with different reaction temperatures. We have accomplished each reaction individually; every step product was isolated and washed with copious amounts of water and dried in an oven at 50 °C for 12 h, and then utilized as a second-step precursor material, and vice versa, to obtain the final composite. Thus, the process was tedious and time-consuming. Eventually, the similarity of the all three reaction’s acetic condition triggered us to optimize this process as a simple one-pot synthesis. As a result, two parallel reactions were carried out under the same conditions, one for sample characterization in each tandem step (diazotization, reduction, and polymerization), and another for straightforward one-pot synthesis. Finally, we successfully achieved the high-performance RGO-PANI nanomaterials in a one-pot synthesis.
Experimental details
Materials
Graphite (< 20 µm), hydrogen peroxide (30%), sodium nitrite (assay 99.9%), and ammonium persulfate (≥ 98%) were purchased from Sigma-Aldrich. Potassium permanganate (assay 99%), sodium nitrate (assay 99%), and hydrochloric acid (36–38%) were obtained from J.T. Baker. 4-Nitroaniline (assay ≥ 98%), tin (II) chloride dihydrate (assay 98%), and aniline (assay ≥ 99.5%) were obtained from Alfa Aesar. The active carbon 99.95% (2–12 µm) was purchased from Sigma-Aldrich. All the other reagents were of analytical purity and used without further purification. Deionized water was used throughout the experiments.
Synthesis
The required amount of graphene oxide (GO) precursor material was prepared by well-known modified Hummer’s methods (Chen et al. 2013; Dreyer et al. 2010; Marcano et al. 2010). Simple one-pot synthesis successfully synthesized the covalently functionalized RGO-PANI nanocomposite. Briefly, the well-sonicated dispersion of 500 mg of GO in distilled water was taken in a round-bottom flask fitted with a mechanical stirrer (300 RPM) kept under the constant temperature of 0–5 °C. The solution of 4-nitroaniline (1 mol) dissolved in ethanol was charged to the round-bottom flask (RBF) with continuous stirring, and then hydrochloric acid 35% solution (2.5 mol) was added. After that, a solution of NaNO2 (1.2 mol) in water was carefully added dropwise to the reaction mass for diazotization. After three hours of stirring, the reaction temperature was manually allowed to reach room temperature by removing an ice bath. Then, the reaction mass was brought to 80 °C and maintained through a hot oil bath. At 80 °C, a SnCl2.2H2O (10 mol) solution was added, with that temperature maintained up to four hours for efficient reduction. After that, the hot oil bath was replaced with a water bath to bring the reaction mass to room temperature (≤ 25 °C) for aniline polymerization. Then, we added the aniline monomer 10:1 wt% with respect to GO, to complete the polymerization in the presence of ammonium peroxydisulfate with hydrochloric acid (pH ≤ 2). Finally, the reaction mass was filtered through Whatman filter paper fitted with a Buchner funnel under vacuum. The wet solid was washed thoroughly with distilled water, followed by acetone, until the washings were colorless. Finally, the wet solid was collected and dried in an oven at 50 °C for 12 h.
Characterization
The X-ray diffraction (XRD) patterns of the obtained samples were measured using an X-ray diffractometer (PANalytical X’Pert PRO) with Cu radiation (λ = 0.15418 nm) in the 2θ range of 5°–90°. The surface morphology was examined by field-emission scanning electron microscopy (FESEM) (300 kV JOEL H-9500) and transmission electron microscopy (TEM) (JOEL 3010). The surface areas of the as-prepared composites were determined using a BET (Micromeritics ASAP 2020).
Electrochemical measurements
The electrochemical measurements were carried out at room temperature using a CHI 614c electrochemical workstation in two-electrode systems. The galvanostatic charge and discharge were performed using Jiehan 5640 multichannel electrochemical workstations. The working electrode was made by mixing with RGO-PANI nanocomposite (90 wt%), acetylene black (5 wt%), and polyvinylidene difluoride (PVDF) (5 wt%) in NMP to form a homogeneous slurry. Then, the slurry was coated with a pre-cleaned nickel plate substrate (area 1 cm2) and dried at 60 °C for 4 h in a vacuum oven. The supercapacitor fabrication was effectively achieved by assembling the above-mentioned RGO-PANI nanocomposites-coated plate as an anode and active carbon plate as a cathode, separated by polypropylene polymer separator in 1 M Na2SO4 gel electrolyte (Unnikrishnan et al. 2016). The cyclic voltammetry (CV) curves were performed in a potential ranging from − 1.0 to 1.0 V at different scan rates from 1 to 100 mV s−1. The galvanostatic charge and discharge measurements were performed using the same potential range (− 1 to + 1) at 0.01 A for stability studies. The electrochemical impedance spectroscopy (EIS) tests were carried out with an AC voltage of five mV in the frequency range of 0.01–105 Hz.
Results and discussion
Characterization of the grafted GO and RGO samples
The grafting of 4-nitroaniline (4NA) on GO was confirmed through both UV and FTIR spectra. The pristine GO and 4NA are showing a distinct absorption band in UV at 231 and 371 nm, respectively (Bertinelli et al. 1977; Konios et al. 2014). In 4NA, the UV shoulder band raised at 340 nm was attributed to the n–π* transition of highly electron-withdrawing NO2 group. Meanwhile, the grafted nitro-GO composite did not possess strong shoulder band in UV spectra shown in (Fig. 1); also, the red shift indicated the successful grafting on the surface of the GO functional groups. After that, effective nitro group reduction was performed with acidic stannous chloride at 80 °C, and the sample was monitored in UV spectra, showing the complete disappearance of nitro shoulder band at 371 nm, which confirmed the conversion of nitro to the amino functional group in the grafted phenyl ring. Furthermore, each composite was used to record the FTIR spectra with the specific range of 400–2000 cm−1 to confirm the reduction process through inherent strong nitro group stretching frequencies of 1560 and 1347 cm−1 (Yadav et al. 2007). In Fig. 2a, the stretching frequencies of the strong nitro group in the nitro composite, which means 4NA grafted GO was diminished entirely after the reduction. It reveals that the process of both grafting and reduction had proceeded successfully. Also, another notable factor in this stannous chloride reduction is that the use of SnCl2 not only reduced the nitro group in the grafted phenyl ring, but also reduced the free oxygen functionalities presented in the both basal and edge planes of GO, resulting in the free amino-phenyl-grafted reduced graphene oxide nanocomposite.

UV–visible spectral data of the grafting and reduction of 4-nitro aniline on graphene oxide

a FTIR data of the grafted and reduced 4-nitro aniline on graphene oxide; b FTIR spectra of the GO, RGO, and RGO-PANI nanocomposites
Characterization of the RGO-PANI nanocomposites
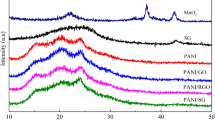
Figure 2b depicts the FTIR spectra of the as-prepared final one-pot RGO-PANI nanocomposite, from which we concluded the covalent functionalization of PANI through a polymerization reaction. The first stretching frequency of N = Q=N arising at 1104 cm−1 reveals the successful covalent functionalization of RGO-PANI (Kumar 2012). Furthermore, we have characterized the crystalline nature and order of the prepared nanocomposites through XRD measurements (Fig. 3). From the XRD data, the sharp diffraction peak at 12° in GO caused by 002 planes shifted at 25° in RGO due to the stacked Sp2− layers of partially removed oxygen functionalities which contained reduced arrays of graphene oxide (Feng et al. 2013; Palanisamy et al. 2013; Velusamy et al. 2017). However, the as-prepared PANI-RGO nanocomposites having the same XRD planes along with various 2θ diffraction peaks at 15°, 21°, 25° indicated the amorphous nature of the composites, as well as the nanometric properties of the RGO-PANI composite. The Raman peaks at 1360 and 1580 cm−1, discretely called D-band and G-band, are shown in Fig. 4. It is well established that the Raman D-band and G-band arise due to the structural disorder of oxygenated Sp3 carbon and E2g phonon of Sp2-hybridized carbon–carbon double bond (Kudin et al. 2008; Matthews et al. 1999). The ID/IG ratio of GO, RGO, and RGO-PANI is 0.9584, 1.2022, and 0.9957, respectively. From ID/IG ratio, it is clear that the disorderliness of pure RGO is reduced after the covalent functionalization of PANI.

XRD data of the GO, RGO, and RGO-PANI composites

Raman data of the GO, RGO, and RGO-PANI composites
Surface morphology and porosity
The surface morphology of the as-prepared RGO-PANI nanocomposites and pristine GO is shown in Fig. 5. The portrayed FESEM images of the pristine GO exhibited a sheet-like morphology with shrinkage, and the RGO-PANI nanocomposites show nanorods-like surface morphology. The covalent functionalization of PANI on RGO sheets through simple sol–gel approach does not interfere in its nanometric structure at the core level. The displayed nanorod-like morphology is self-assembly of PANI on the supportive RGO nanosheets. The oriented assembly of this nanostructure enhances the conductivity of the PANI as well as RGO (Krukiewicz and Katunin 2016; Mokhtar et al. 2017). In Fig. 6, high-resolution TEM images of RGO-PANI are shown in 1, 0.2 µm, 100, and 50 nm. It was evident that the PANI nanorods are uniformly oriented on the atomic thick RGO nanosheets. The grain boundaries shown in the TEM pictures also proved their functionalization. Furthermore, the specific textural properties of the RGO-PANI nanocomposite were elucidated with the help of TEM in different magnification. Figure 7 depicts the PANI nanorods accumulated on the nanosheets of the curved RGO. The lattice space of the RGO and PANI was measured in 5-nm scale in which the lattice of RGO 0.245 nm is smaller than the PANI 0.272 nm. The porosity and the surface area of the RGO-PANI nanocomposites were monitored by the Brunauer–Emmett–Teller method, displayed in Fig. 8, in which, the as-prepared nanocomposites show the best surface area among the pristine PANI and RGO. The surface area of the RGO-PANI, PANI, and RGO was 35.75, 19.20, and 7.79 m2g−1. The BET surface area of the RGO-PANI composite is higher than that of both pristine RGO and PANI because the curved surface of the RGO nanosheets is covalently functionalized with nanorod-like PANI. The surface of RGO tightly bound with PANI and wrapped over the nanosheets, which synergistically, increases the pores on the composite material and leads to the increased porosity and surface area of the composite.

FESEM images of GO, RGO, and RGO-PANI composites

TEM images of RGO-PANI composite in different magnification

TEM images of RGO-PANI composite in higher resolution

BET data of the GO, RGO, and RGO-PANI composites
Electrochemical properties of the samples
The electrochemical properties of the as-prepared covalently functionalized RGO-PANI nanocomposite were investigated through cyclic voltammetry (CV) with fabricated supercapacitor package in the two-electrode system, as depicted in Fig. 9a. The potential varied linearly from − 1.0 to + 1.0 V with different scan rates starting from 1 mV to 100 mV, while the CV curves of active redox peak, increasing with the scan rate, clearly indicates the stable electrochemical activity of the RGO-PANI nanocomposites. However, the quasi-rectangular-shaped CV curves demonstrate the dominant double-layer (electrostatic) capacitance of carbonaceous material, as compared to the available redox-active PANI counterparts present in the RGO-PANI nanocomposites. That indicates the PANI has held the conductivity region in synergistic RGO-PANI nanocomposites. The specific capacitance of the as-prepared RGO-PANI nanocomposite is calculated with Eq. (1):

a CV of the RGO-PANI composite electrodes at various scan rates (1–100 mV) in 1 M Na2SO4 solution; b GCD of the RGO-PANI composite at different current density at constant potential range of 0.0–2.0 V; c cyclic stability test of the RGO-PANI composite at constant potential of 2 V and 10 mA constant current density; d EIS of the RGO and RGO-PANI composite
From the above equation, the integral I(V) dV is the active electrochemical area from CV, m is a total mass of two-electrode (positive and negative) material, ν is the scan rate, and ΔV is the potential (V). The lowest 1 mV/s scan rate electrochemical active area was taken to calculate the single-electrode specific capacitance. Various reports of carbon–polyaniline-based supercapacitor electrode materials were matched with our study, and the compared results are shown in Table 1. The prepared covalently functionalized RGO-PANI shows the highest specific capacitance of 490 F g−1. As a result, the as-prepared supercapacitor was investigated with the galvanostatic charge and discharge measurement with various current densities at a constant potential (2 V). Different current densities, starting from 5 to 25 mA, were carried out to evaluate the specific storage capability of the device in both IR drop and discharge rate, with the results portrayed in Fig. 9b. Finally, the constant potential 2 V and current density 10 mA were chosen to evaluate the cyclic stability. The initial capacitance (calculated from GCD by Δt) of the first cycle was retained up to 10,000 cycles, as shown in Fig. 9c. The electrochemical impedance spectra (EIS) were assigned to investigate the impedance of the as-prepared RGO-PANI material, as depicted in Fig. 9d. Compared to bare RGO, the covalently functionalized RGO-PANI shows low impedance in EIS. The impedance of pristine RGO and RGO-PANI was 29.12 and 26.36 Ω, respectively. The low internal resistance of the functionalized RGO-PANI leads to excellent cycle stability and high power density.
Conclusions
We have developed and demonstrated a promising approach to preparing covalently functionalized RGO-PANI nanocomposites in simple one-pot synthesis with inexpensive commercially available precursors. The proposed strategy through diazotization reaction is a hopeful paradigm for grafting. The RGO-PANI nanocomposite delivered a high specific capacitance of 490 F g−1 at 1 mV s−1 with excellent cyclic stability. Finally, the prepared nanocomposites have outstanding structural stability, due to the robust covalent bonding between RGO and PANI. The covalent functionalization nullified the structural instability and degradation of PANI during the charge–discharge cycles. To the best of our knowledge, the proposed one-pot covalent functionalization of RGO-PANI adopting diazotization grafting strategy has not been reported in any scientific literature. This new approach may open the gates of high-performance nanomaterials in energy storage regimes.
References
Aricò AS, Bruce P, Scrosati B, Tarascon J-M, Van Schalkwijk W (2005) Nanostructured materials for advanced energy conversion and storage devices. Nat Mater 4:366–377
Arjun N, Pan G-T, Yang TC (2017) The exploration of Lanthanum based perovskites and their complementary electrolytes for the supercapacitor applications. Results Phys 7:920–926
Baker CO, Huang X, Nelson W, Kaner RB (2017) Polyaniline nanofibers: broadening applications for conducting polymers. Chem Soc Rev 46:1510–1525
Basnayaka PA, Ram MK (2017) A review of supercapacitor energy storage using nanohybrid conducting polymers and carbon electrode materials. In: Kalia S (ed) Conducting polymer hybrids. Springer, Berlin, pp 165–192
Behera SR, Dash DP (2017) The effect of urbanization, energy consumption, and foreign direct investment on the carbon dioxide emission in the SSEA (South and Southeast Asian) region. Renew Sustain Energy Rev 70:96–106
Bertinelli F, Palmieri P, Brillante A, Taliani C (1977) Electronic-excited states of nitroanilines. II. A configuration interaction study and UV spectrum of the paranitroaniline single crystal. Chem Phys 25:333–341
Borenstein A, Hanna O, Attias R, Luski S, Brousse T, Aurbach D (2017) Carbon-based composite materials for supercapacitor electrodes: a review. J Mater Chem A 5(25):12653–12672
Chen J, Yao B, Li C, Shi G (2013) An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon 64:225–229
Chen J et al (2016) The origin of improved electrical double-layer capacitance by inclusion of topological defects and dopants in graphene for supercapacitors. Angew Chem Int Ed 55:13822–13827
Chen F, Wan P, Xu H, Sun X (2017a) Flexible transparent supercapacitors based on hierarchical nanocomposite films. ACS Appl Mater Interfaces 9(21):17865–17871
Chen L-F, Lu Y, Yu L, Lou XWD (2017b) Designed formation of hollow particle-based nitrogen-doped carbon nanofibers for high-performance supercapacitors. Energy Environ Sci 10(8):1777–1783
Dai W et al (2016) Fabrication of sandwich nanostructure graphene/polyaniline hollow spheres composite and its applications as electrode materials for supercapacitor. Mater Res Bull 76:344–352
Dong L, Xu C, Li Y, Huang Z-H, Kang F, Yang Q-H, Zhao X (2016) Flexible electrodes and supercapacitors for wearable energy storage: a review by category. J Mater Chem A 4:4659–4685
Dreyer DR, Park S, Bielawski CW, Ruoff RS (2010) The chemistry of graphene oxide. Chem Soc Rev 39:228–240
Eng AYS, Chua CK, Pumera M (2016) Facile labeling of graphene oxide for superior capacitive energy storage and fluorescence applications. Phys Chem Chem Phys 18:9673–9681
Feng H, Cheng R, Zhao X, Duan X, Li J (2013) A low-temperature method to produce highly reduced graphene oxide. Nat Commun 4:1539. https://doi.org/10.1038/ncomms2555
Gao Z et al (2014) Chemically grafted graphene-polyaniline composite for application in supercapacitor. Electrochim Acta 133:325–334. https://doi.org/10.1016/j.electacta.2014.04.033
González A, Goikolea E, Barrena JA, Mysyk R (2016) Review on supercapacitors: technologies and materials. Renew Sustain Energy Rev 58:1189–1206
Guo B et al (2017) Redox-active organic molecules functionalized nitrogen-doped porous carbon derived from metal-organic framework as electrode materials for supercapacitor. Electrochim Acta 223:74–84
Hong X, Zhang B, Murphy E, Zou J, Kim F (2017) Three-dimensional reduced graphene oxide/polyaniline nanocomposite film prepared by diffusion-driven layer-by-layer assembly for high-performance supercapacitors. J Power Sources 343:60–66
Hwang M, Oh J, Kang J, Seong K-D, Piao Y (2016) Enhanced active sites possessing three-dimensional ternary nanocomposites of reduced graphene oxide/polyaniline/Vulcan carbon for high-performance supercapacitors. Electrochim Acta 221:23–30
Konios D, Stylianakis MM, Stratakis E, Kymakis E (2014) Dispersion behavior of graphene oxide and reduced graphene oxide. J Colloid Interface Sci 430:108–112
Krukiewicz K, Katunin A (2016) The effect of reaction medium on the conductivity and morphology of polyaniline doped with camphor sulfonic acid. Synth Met 214:45–49
Kudin KN, Ozbas B, Schniepp HC, Prudomme RK, Aksay IA, Car R (2008) Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett 8:36–41
Kumar NA (2012) Polyaniline-grafted reduced graphene oxide for efficient electrochemical supercapacitors. ACS Nano 6:1715–1723. https://doi.org/10.1021/nn204688c
Kumar M et al (2013) Synthesis and characterization of covalently-grafted graphene–polyaniline nanocomposites and its use in a supercapacitor. Chem Eng J 231:397–405. https://doi.org/10.1016/j.cej.2013.07.043
Li W, Yao H, Zhang G, Yang Y (2017) A Ni/Zn bi-metallic coordination supramolecular network applied for high-performance energy storage material. Electrochim Acta 228:233–240
Ma H, He J, Xiong D-B, Wu J, Li Q, Dravid V, Zhao Y (2016) Nickel cobalt hydroxide@ reduced graphene oxide hybrid nanolayers for high-performance asymmetric supercapacitors with remarkable cycling stability. ACS Appl Mater Interfaces 8:1992–2000
Marcano DC et al (2010) Improved synthesis of graphene oxide. ACS Nano 4(8):4806–4814
Matthews M, Pimenta M, Dresselhaus G, Dresselhaus M, Endo M (1999) Origin of dispersive effects of the Raman D band in carbon materials. Phys Rev B 59:R6585
Mokhtar N, Chye DAT, Phang SW (2017) Morphology, conductivity and microwave absorption behavior of polyaniline nanocomposites after chemical treatment. Polym Polym Compos 25:545
Mondal S, Rana U, Malik S (2017) Reduced graphene oxide/Fe3O4/polyaniline nanostructures as electrode materials for an all-solid-state hybrid supercapacitor. J Phys Chem C 121:7573–7583
Mousavi MF, Hashemi M, Rahmanifar MS, Noori A (2017) Synergistic effect between redox additive electrolyte and PANI-rGO nanocomposite electrode for high energy and high power supercapacitor. Electrochim Acta 228:290–298
Palanisamy S, Chen S-M, Sarawathi R (2012) A novel nonenzymatic hydrogen peroxide sensor based on reduced graphene oxide/ZnO composite modified electrode. Sens Actuators B Chem 166:372–377
Palanisamy S, Ku S, Chen S-M (2013) Dopamine sensor based on a glassy carbon electrode modified with a reduced graphene oxide and palladium nanoparticles composite. Microchim Acta 180:1037–1042
Palanisamy S et al (2017a) A novel laccase biosensor based on laccase immobilized graphene-cellulose microfiber composite modified screen-printed carbon electrode for sensitive determination of catechol. Sci Rep 7:41214
Palanisamy S et al (2017b) Synthesis and characterization of polypyrrole decorated graphene/β-cyclodextrin composite for low level electrochemical detection of mercury (II) in water. Sens Actuators B Chem 243:888–894
Salunkhe RR et al (2014) Fabrication of symmetric supercapacitors based on MOF-derived nanoporous carbons. J Mater Chem A 2:19848–19854
Salunkhe RR, Young C, Tang J, Takei T, Ide Y, Kobayashi N, Yamauchi Y (2016) A high-performance supercapacitor cell based on ZIF-8-derived nanoporous carbon using an organic electrolyte. Chem Commun 52:4764–4767
Sumboja A, Foo CY, Wang X, Lee PS (2013) Large areal mass, flexible and free-standing reduced graphene oxide/manganese dioxide paper for asymmetric supercapacitor device. Adv Mater 25:2809–2815
Sun G et al (2014) Fabrication of ultralong hybrid microfibers from nanosheets of reduced graphene oxide and transition-metal dichalcogenides and their application as supercapacitors. Angew Chem 126:12784–12788
Sun H, She P, Xu K, Shang Y, Yin S, Liu Z (2015) A self-standing nanocomposite foam of polyaniline@ reduced graphene oxide for flexible super-capacitors. Synth Met 209:68–73
Takahashi A et al (2017) Effect of evacuation on liver function after the Fukushima Daiichi Nuclear Power Plant accident: the Fukushima Health Management Survey. J Epidemiol 27:180–185
Tayel MB, Soliman MM, Ebrahim S, Harb ME (2016) Sprayed polyaniline layer onto chemically reduced graphene oxide as electrode for high performance supercapacitor. Synth Met 217:237–243
Unnikrishnan B, Wu C-W, Chen I-WP, Chang H-T, Lin C-H, Huang C-C (2016) Carbon dot-mediated synthesis of manganese oxide decorated graphene nanosheets for supercapacitor application. ACS Sustain Chem Eng 4:3008–3016
Velusamy V, Palanisamy S, Chen S-M, Chen T-W, Selvam S, Ramaraj SK, Lou B-S (2017) Graphene dispersed cellulose microfibers composite for efficient immobilization of hemoglobin and selective biosensor for detection of hydrogen peroxide. Sens Actuators B Chem 252:175–182
Wang G, Zhang L, Zhang J (2012a) A review of electrode materials for electrochemical supercapacitors. Chem Soc Rev 41:797–828
Wang H, Maiyalagan T, Wang X (2012b) Review on recent progress in nitrogen-doped graphene: synthesis, characterization, and its potential applications. ACS Catal 2:781–794
Wu W, Li Y, Yang L, Ma Y, Pan D, Li Y (2014) A facile one-pot preparation of dialdehyde starch reduced graphene oxide/polyaniline composite for supercapacitors. Electrochim Acta 139:117–126
Xia X et al (2014) Synthesis of free-standing metal sulfide nanoarrays via anion exchange reaction and their electrochemical energy storage application. Small 10:766–773
Xia C, Jiang Q, Zhao C, Beaujuge PM, Alshareef HN (2016) Asymmetric supercapacitors with metal-like ternary selenides and porous graphene electrodes. Nano Energy 24:78–86
Xiong C et al (2017) Two-step approach of fabrication of interconnected nanoporous 3D reduced graphene oxide-carbon nanotube-polyaniline hybrid as a binder-free supercapacitor electrode. J Alloys Compd 695:1248–1259
Yadav B, Ali I, Kumar P, Yadav P (2007) FTIR and laser Raman spectra of 2-hydroxy-5-methyl-3-nitropyridine. Indian J Pure Appl Phys 45:979–983
Yamamura H, Yaguchi S, Itoh K (2017) The new radiation emergency medical system in Japan: lessons from the Fukushima Nuclear Plant Accident. Prehospital Disaster Med 32:S4–S4
Yu P, Zhao X, Huang Z, Li Y, Zhang Q (2014) Free-standing three-dimensional graphene and polyaniline nanowire arrays hybrid foams for high-performance flexible and lightweight supercapacitors. J Mater Chem A 2:14413–14420
Yu N, Yin H, Zhang W, Liu Y, Tang Z, Zhu MQ (2016) High-performance fiber-shaped all-solid-state asymmetric supercapacitors based on ultrathin MnO2 nanosheet/carbon fiber cathodes for wearable electronics. Adv Energy Mater 6:1501458
Yuan C, Wu HB, Xie Y, Lou XWD (2014) Mixed transition-metal oxides: design, synthesis, and energy-related applications. Angew Chem Int Ed 53:1488–1504
Zhang J, Jiang J, Li H, Zhao XS (2011) A high-performance asymmetric supercapacitor fabricated with graphene-based electrodes. Energy Environ Sci 4:4009. https://doi.org/10.1039/c1ee01354h
Zhong C, Deng Y, Hu W, Qiao J, Zhang L, Zhang J (2015) A review of electrolyte materials and compositions for electrochemical supercapacitors. Chem Soc Rev 44:7484–7539
Acknowledgements
The authors thank the Centre for Precision Analysis and Research, National Taipei University of Technology, Taipei, for financial support for this research. Also, the authors want to thank Professor Chaochin Su, Department of molecular science and engineering, National Taipei University of Technology, Taipei, for assistance in the electrochemical stability tests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Arjun, N., Uma, K., Pan, GT. et al. One-pot synthesis of covalently functionalized reduced graphene oxide–polyaniline nanocomposite for supercapacitor applications. Clean Techn Environ Policy 20, 2025–2035 (2018). https://doi.org/10.1007/s10098-018-1573-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10098-018-1573-8