
Abstract
Cystic fibrosis (CF) is a complex inherited disease which affects many organs, including the pancreas and liver, gastrointestinal tract and reproductive system, sweat glands and, particularly, the respiratory system. Pseudomonas aeruginosa is the main cause of chronic airway infection. In order to reduce morbidity and mortality due to lung infection by P. aeruginosa, aerosol antibiotics have been used to achieve high local concentrations in the airways and to reduce systemic toxicity. In the course of this review, the current treatments to control CF lung infections by P. aeruginosa are presented. Some innovative aerosol formulations such as liposomes and microspheres are herein reviewed, which may improve the efficiency of anti-pseudomonal agents, and ensure patients’ compliance to treatments, by reducing dosing frequency and/or drug dose, while maintaining therapeutic efficacy, preventing the occurrence of bacterial resistance and/or reducing adverse effects due to their controlled-release properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction to cystic fibrosis disease
Cystic fibrosis (CF) is an inherited, recessive and autosomal disease which affects approximately one in 2,000 newborns in Caucasian populations. It is, however, found in all ethnic groups [1].
This disorder is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7. A deletion of phenylalanine in the amino acid position 508 is the most common mutation [2], but more than 800 different mutations have been identified [1, 3]. In addition, CF is a multi-functional disease in which several organs are affected, including the pancreas and liver, gastrointestinal tract and reproductive system, sweat glands and, particularly, the respiratory system [1, 4–6]. Some carriers of a single CFTR mutation suffer from some pulmonary and gastrointestinal symptoms, but not at a high level [4]. This review will focus on respiratory symptoms caused by this disease. Because the CFTR protein regulates ion and water movement across the epithelium, these mutations lead to malfunction of the chloride channel in CF patients [1, 3, 4, 7]. Thus, the scenario includes decreased chloride secretion into the airways and increased sodium absorption from the airways that lead to relative dehydration of the airway mucus [8]. Therefore, the volume of the airway surface liquid (ASL) decreases, leading to the accumulation of purulent secretions (expectorated form is known as sputum) [4] and impaired mucociliary clearance [9, 10]. Mucociliary clearance is an important defence mechanism against pathogens and foreign substances, but this mechanism is reduced in CF patients [4, 11–13], leading to chronic infections [10, 14]. On one hand due to the reduced mucociliary clearance in these patients, it is less of a challenge to inhalational drugs delivery, but on the other hand, mucociliary clearance could be improved when mucus-thinning agents are co-administered [4]. In addition, from in vitro studies, Coakley et al. proposed that mucus adherence to epithelial surfaces may be strengthened by the low pH of ASL because of absent CFTR-dependent HCO3 − secretion [15].
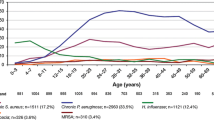
This review will focus on Pseudomonas aeruginosa, which is the main pathogen that leads to chronic CF lung disease: 54.4 % of the whole CF patient population are infected with this bacterium, which is found in 80 % of the patients by the age of 18 years [16]. P. aeruginosa predominates in the majority of adult CF patients [7, 17], whereas Staphylococcus aureus (including methicillin-resistant S. aureus, MRSA) [18] and Haemophilus influenzae are the main pathogens in children [7, 10, 19, 20], as shown in Fig. 1. However, it should be kept in mind that other pathogens, such as Burkholderia spp., Achromobacter spp. and Stenotrophomonas maltophilia also contribute to morbidity and mortality [1, 14, 21]. Recent studies have shown that the organisms belonging to the Streptococcus milleri group may also be an intrinsic component of the CF airway microbiome and have recently been implicated as aetiological agents of pulmonary exacerbations in adult CF patients [22]. Besides, fungal and viral infections also play an important role in the development of CF lung disease [17]. In addition, the bacterial community in these patients appears to be polymicrobial in nature, including several anaerobic species, such as the genera Prevotella, Veillonella, Propionibacterium and Actinomyces. These anaerobic bacteria are clinically relevant for CF lung disease and consist in a complex issue which is currently under investigation. The clinical significance of these species remains unclear and it is the main reason why clinicians have not specifically treated anaerobes. Nevertheless, some in vitro data suggest that P. intermedia may contribute to lung disease in CF patients, which, if demonstrated in vivo, may facilitate the decision for antibiotic therapy [17, 23]. Culture-independent microbial profiling methods showed that microbial communities in the airways of CF patients are complex ecosystems with high microbial diversity. Such new methods revealed the infraspecific diversity in Candida albicans, C. parapsilosis and Aspergillus fumigates, and cryptic and new unculturable (or difficult to grow in vitro) species, most of them described as human pathogens. They also revealed the complex interaction between typical pathogens and microbiota, such as the association between P. aeruginosa and anaerobes. P. aeruginosa can grow under reduced oxygen tension (as described next), as well as other anaerobic bacteria, and C. albicans can also grow under anaerobic conditions [24]. Microbial community diversity was shown to evolve with patients’ aging. Young CF patients who show less airway inflammation and better pulmonary function possess the greatest diversity of airway bacterial species (including anaerobic community). With aging, the diversity of the microbial communities tends to be substantially reduced [24, 25], which might be linked to the gradual decline in the lung function, as suggested by some investigators [26, 27]. Some authors suggest that antibiotic treatments contribute to the loss of airway microbiome diversity [27]. Still controversial and requiring further investigation, the management and preservation of the diversity of the lung microbial communities might be a way to prolong lung functions and life expectancies in CF patients, and require new treatment strategies beyond the scope of the present review [25, 26].

Prevalence of predominate bacterial infections in cystic fibrosis (CF) patients according to age. Reproduced from the Cystic Fibrosis Foundation patient registry, annual data report 2011 [150], with permission
Infection with P. aeruginosa in CF patients
Development of chronic infection
Geller characterises the lung disease in CF as an endobronchial infection, exaggerated inflammatory response, progressive airway obstruction, bronchiectasis and eventual respiratory failure [16]. P. aeruginosa is generally considered as the most important pathogen associated with CF in terms of prevalence and pathogenicity, and this opportunistic bacterium is the main pathogen leading to advanced CF lung disease [14, 16, 19]. P. aeruginosa is a Gram-negative bacillus (non-capsulate and non-sporing) that affects particularly the lower respiratory tract [28]. Without any treatment, P. aeruginosa infection persists in spite of the recruitment of the host’s defence mechanisms and leads to decreased respiratory function [14], and most patients die at a young age of lung complications. Under intensive treatment, the mean expected lifetime of CF patients is >35 years, and in some centres, it is >50 years [1].
P. aeruginosa is transmitted by a direct contact between carriers and via environmental reservoirs. The initial infection usually involves a planktonic, non-mucoid strain of P. aeruginosa, which is able to penetrate the mucus by its flagellar activity [2]. The initial infection is not associated with an immediate and rapid decline in lung function, and early P. aeruginosa isolates appear non-resistant to the antibiotic treatment [16], giving an opportunity for an early efficient therapeutic intervention [29]. Aggressive and early antibiotic treatments of P. aeruginosa infection were shown to increase the life expectancy of CF patients [16, 19]. Transmission between infected CF patients may occur and be favoured in communities like day hospitals, wards where CF patients come into contact with each other, leading to replacement, under antimicrobial pressure, of initially acquired more antibiotic-sensitive clones with resistant ones [30]. In addition, P. aeruginosa was shown to persist and to multiply in hospital equipments and bathroom sinks, which suggests that high hygienic standards should be applied in order to minimise the risk of cross-infection [31]. After the initial infection, there follows a variable period of “intermittent or transient infection”, which refers to patients with less than 50 % of the monthly cough swab or sputum cultures found to be positive for P. aeruginosa over a 12-month period, according to the Leeds classification [32]. If left untreated, the starting infection eventually evolves towards a “chronic infection” which, according to the Leeds classification, refers to patients with more than 50 % of the monthly cultures found to be positive over a 12-month period [32]. Chronic infection involves mucoid strains of P. aeruginosa, which synthesises an exopolysaccharide (alginate) matrix or biofilm that protects bacteria against antibiotics [10] and phagocytosis [3, 16, 29], as shown in Fig. 2 (e.g. it may be difficult for neutrophils to penetrate into mucus plaques). Though being aerobic, P. aeruginosa within biofilm is able to survive in the hypoxic environment of the CF mucus plug, which diminishes its sensitivity to antibiotics [2]. In most cases, mucoid P. aeruginosa infections cannot be eradicated.

Macrocolonies in chronic infection by Pseudomonas aeruginosa which resist to innate defences, including neutrophils. Reproduced from reference [2], with permission
Early aggressive antibiotic treatment, which is crucial to prevent or at least to delay chronic lung colonisation, implies the early diagnosis of initial or recurrent contaminations. P. aeruginosa is diagnosed in sputum, hypopharyngeal or endolaryngeal suctions, or deep throat culture [3, 10]. Routine bacterial cultures appear to have limited sensitivity for detecting the initially very low numbers of bacterial cells. Several molecular assays, generally based on the polymerase chain reaction (PCR) technique, and especially on its quantitative variant, have, therefore, been proposed for the early detection of P. aeruginosa and for confirming the respective eradication after antibiotic treatment in young CF patients, but they still require investigation of their clinical significance and relevance before implementation in routine laboratories [33, 34].
Resistance to antibiotic therapy
Currently, antibiotic resistance is increasing and we need new strategies to overcome this reality. In spite of intensive antibiotic therapy and defence mechanisms of the immune system, P. aeruginosa has the ability to survive and persist for years in the CF patient’s lungs, owing to its adaptive mechanisms [1, 3, 35, 36]. P. aeruginosa has the ability to form biofilms [37], develop a mucoid phenotype and lack membrane porins (that are important to antibiotic diffusion) [4, 38–40], or may develop an active drug efflux mechanism [4, 10, 19, 41].
Additionally, different antibiotics display different modes of action and, therefore, different “rates of resistance”: (a) beta-lactams (e.g. carbapenems and the monobactam aztreonam) interfere with bacterial cell wall synthesis [3, 28]; (b) quinolones inhibit DNA synthesis by acting on topoisomerases, particularly topoisomerase II (DNA gyrase) and IV (which are essential for maintenance of the appropriate DNA topological state for replication and transcription) [42, 43]; and (c) aminoglycosides (e.g. tobramycin, amikacin and gentamicin) [3, 28] inhibit protein synthesis by binding bacterial ribosomal subunits or inhibiting ribosomal enzymes [3].
As described by Grégoire et al., P. aeruginosa becomes resistant to fluoroquinolones through two known mechanisms: drug efflux pumps that reduce the accumulation of antibiotics in the cell and point mutations in the genes of the quinolone target enzymes, DNA gyrase and topoisomerase IV, owing to amino acid substitutions in the corresponding genes gyrA or gyrB (for DNA gyrase) and parC or parE (for topoisomerase IV) [42, 43]. According to some authors, fluoroquinolones should be used prudently to avoid further resistance. In addition, this class of antibiotics is associated with cross-resistance to other antibiotics (e.g. aminoglycosides) and act as selectors for MRSA [18]. With respect to other antibiotic classes, P. aeruginosa resists aminoglycosides mainly by reducing their transport through the membrane [44] and reduces the beta-lactam activity by some mechanisms as described in Table 1. Recently, a study concluded that resistance to tobramycin in P. aeruginosa isolates from CF patients under antimicrobial therapy may occur, while colistin resistance appears to be rare [45].
As reported by Ibrahim et al., the strategy may rely on the combination of antibiotics to avoid antibiotic resistance. Recently, some inhalable particles co-encapsulating two or more antibiotics displayed good results, especially when the microorganism is difficult to kill with one antibiotic [4, 46]. One strategy consists in encapsulating the drugs in liposomes or microspheres, as will be discussed in this review [4]. When the resistance is due to the drug efflux mechanism, the alternative consists in the administration of efflux pump inhibitors [19]. With respect to CF, compounds such as broad-spectrum efflux pump inhibitors in P. aeruginosa are in clinical development for use as an aerosol [19, 47]. A study has recently evaluated the efficacy of efflux pump inhibitors in reducing ciprofloxacin and levofloxacin minimum inhibitory concentrations (MICs), which concluded that the activity of these inhibitors cannot be generalised to all bacteria and to all antibiotics of the same class [47].
In what pertains to inhaled antibiotics, there are areas of variable antibiotic concentrations in the airways. The low concentration areas allow the selection of more resistant microbes. Alternating months of antibiotics reduce the selective pressure on the bacteria, and it is necessary to understand whether “drug holidays” or alternating chronic antibiotics may prevent the emergence of multi-resistant organisms in CF. Resistance may be transient and reverted when antibiotic selective pressure is removed, a process known as adaptive resistance [3]. According to some authors, there is no present definition of in vitro resistance relating to the use of aerosolised formulations [19, 48] because the analyses are related to systemic administration and not to high concentrations achievable in the pulmonary tract [14].
Pharmacokinetics and pharmacodynamics
Recently, some authors indicated that the MIC criterion is insufficient to choose a drug, taking into account certain aspects such as the individual variation in drug metabolism and clearance, and, also, the mechanism of antimicrobial killing.
The time over MIC (T > MIC), the ratio of peak concentration to the MIC (Cmax/MIC) and the ratio of the area under the concentration-versus-time curve to the MIC (AUC/MIC) are usually the parameters used to predict the antibacterial activity. The last two parameters are the mostly used for fluoroquinolones [43].
Grégoire et al. studied a pharmacokinetic–pharmacodynamic (PK-PD) model to describe the effects of ciprofloxacin against P. aeruginosa in vitro and concluded that MIC is inadequate to assess the susceptibility of a bacterial strain leading to regrowth during time–kill studies. However, the PK-PD model may be used to compare antibiotic efficacies for different bacterial strains and to optimise the dose in order to prevent the emergence of resistance [42]. Measures other than the MIC, such as minimum eradication concentration, have not been established, but studies are ongoing [14].
Aminoglycosides and fluoroquinolones have a “concentration-dependent” effect [16]. Thus, the AUC/MIC ratio is associated with the extent of bacterial killing. High Cmax/MIC ratios can suppress the selection of resistant organisms for antibiotics [14]. However, concentration-dependent killing may promote resistance, because of the prolongation of the residence time in the airway at sub-MIC levels (toward the end of a long dosing interval) [16]. Nevertheless, some studies demonstrated no statistical difference in the emergence of resistance between the high- and low-dose arms, highlighting the need for more studies in this area [43].
Technologies that increase residence time in the airways make perfect sense for beta-lactams (time-dependent killing). In contrast to aminoglycosides and fluoroquinolones, beta-lactams have a “time-dependent” effect [16]. Thus, the percentage of a dosing interval in which concentrations exceed the MIC is linked to antimicrobial effects and more frequent dosing is required in order to keep the lung concentration above the MIC (% t > MIC) [14]. Maintaining the concentration at a given multiple above the MIC for longer portions of the dosing interval is associated with better antibacterial effect, but increasing the concentration above this multiple does not improve the killing effect [49].
Difficulties in the interpretation of antibiotic concentrations
In order to prevent antibiotic resistance by bacteria, such as P. aeruginosa, antibiotics need to be administered taking into account PK and PD parameters. The common method is to measure the concentrations in tissue homogenates (e.g. lung homogenate) and to compare these with measures derived from the corresponding blood samples. However, tissues are made of distinct compartments in which the drug may not be homogeneously distributed. Therefore, the concentrations obtained may not represent the (active) concentration of the antibiotic at the site of infection [50, 51]. Thus, efforts have been made to measure the concentration of antibiotics at infection sites (e.g. lungs) because the distribution of antibiotics is, quite likely, different in a variety of tissues and in the serum [52].
However, it is difficult to determine the antibiotic concentration at the site of the infection. In diseases such as CF, antibiotic concentration in the epithelial lining fluid (ELF) could represent antibacterial activity. This fluid is measured on the interior surface of the alveolar wall, which is a component of the blood–alveolar barrier. This barrier is composed of another membrane, the capillary wall. These two “walls” are separated by a fluid-filled interstitial space. Regarding these characteristics, antibiotics measured in the ELF need to cross these barriers.
However, the ratios of ELF to plasma concentrations are different between antibiotics. The reasons are not clarified, but it is known that some confounding factors may affect the measures. The confounding factors may include physicochemical characteristics intrinsic to the molecules and technical factors or errors in the method of measurement (e.g. bronchoalveolar lavage, BAL). ELF is a mixture of components, including cells, which may be lysed during the measurement and it could artificially increase or decrease the measured antibiotic concentration. These “unreal” results are related to the concentration of the antibiotic in cells and in serum [52]. This error will vary with the antibiotic concentration in cells and with the number of cells in the ELF.
Technical errors exist, such as:
-
(a) Correction of the volume of ELF sampled by BAL and the amount of antibiotic contained in the sample for drug-free saline during the BAL procedure. This correction is frequently performed with urea because it is used as an endogenous marker that can easily cross the membranes. Thus, it is assumed that the concentration of urea in the ELF is the same as the serum concentration.
-
(b) The “dwelling time” of fluid during the measurement method because some additional urea appear to diffuse from some tissues when the dwelling times are prolonged. Thus, the ELF volume may be overestimated [52, 53].
Other factors can affect antibiotic concentrations measured in the ELF. Moreover, some authors developed simulations to estimate ELF concentrations of different antibiotics considering the impact of protein binding, different lipid solubilities and molecular weights, and lysis of cells. However, in lung diseases such as CF, these studies have to be performed taking into account the modifications in the cells, tissues and fluids of these patients [52]. Additionally, the route of administration also has importance in these measurements because antibiotics administered by oral or intravenous routes need to cross the blood–alveolar barrier but inhaled antibiotics do not (when a local effect is claimed).
Although these are interesting approaches, PK/PD parameters are usually expressed taking into account the serum concentrations. Even though they are not deeply discussed, it seems that these concentrations are directly related to microbiological outcomes.
Treatment of patients with CF
Current treatment and some investigational approaches
The aim of CF treatment is to increase the life expectancy of patients and to decrease their symptoms, attenuating disease progression. Currently, antibiotics (such as aminoglycosides, beta-lactams, polymyxins and fluoroquinolones [16]) and anti-inflammatory drugs are used to control the inflammation and infection of the respiratory tract, particularly by P. aeruginosa, as previously remarked. In addition, bronchodilators, mucolytics and osmotic agents are administered to improve sputum and airway clearance [4]. Although these agents will not be discussed in this review, several of their features are particularly interesting: (a) osmolytes are not actively transported and poorly absorbed (e.g. mannitol can restore ASL volume) and (b) inhaled hypertonic saline agents are able to draw water to the airway surface [2, 8]. However, these agents show difficulties in demonstrating efficacy, owing to the short duration of active therapy [2].
In what relates to antibiotics, a number of integrated factors must be taken into consideration to choose the drug and delivery method: pharmacological and pharmacodynamic considerations, toxicity, cost, patients’ characteristics, lung function, symptoms and others [7]. Thus, there is no standard treatment for patients with CF lung disease.
Antibiotics
Although there is no standard treatment, a combination of at least two antibiotics is generally used to treat infection by P. aeruginosa in CF patients, which includes intravenous aminoglycosides (and nebulised tobramycin), intravenous or oral fluoroquinolones, and intravenous beta-lactams [4].
-
Aminoglycosides
Tobramycin is an aminoglycoside which is currently used as an aerosol nebulised antibiotic and accepted as a standard treatment for CF patients [19]. The tobramycin inhalation solution was approved in 1998 (TOBI® Novartis Pharmaceuticals, Basel, Switzerland) [14, 54] with the PARI LC Plus nebuliser [16], and the administration of 300 mg (load) twice daily has been investigated alternating 28-day on/off treatment cycles in order to improve lung function (measured as FEV1—volume exhaled at the end of the first second of forced expiration) and to avoid toxicity (due to systemic absorption) [14]. Recently, a study concluded that the use of tobramycin inhalation solution was associated with significantly reduced mortality among patients with CF [55]. However, the administration time is extended for almost 20 min, which is a problem for patient compliance. Taking into account the development of dry-powder inhalers and sustained-release formulations, this problem may be solved by faster and fewer daily administrations, as described below in this review. In addition, aminoglycosides have their activity impaired due to their low penetration in mucus [16, 19]. This appears to result from the binding of cationic moieties on the aminoglycosides to anionic substances present in the sputum of these patients [14].
-
Fluoroquinolones
Fluoroquinolones are under investigation and may be administered during the “off” cycle of tobramycin inhalation solution to improve patient outcomes and to avoid drug resistance [14]. They are being developed for inhalation use, with particular focus on ciprofloxacin and levofloxacin, as described next in this review [56]. In addition, their antibacterial activity is not impaired by low diffusion into the mucus, in contrast to aminoglycosides [14, 16]. Ciprofloxacin is a second-generation fluoroquinolone and it is one of the most widely available fluoroquinolone antibiotics, with activity against several aerobic Gram-positive and Gram-negative bacteria [57]. Introduced in 1985 as an oral treatment of CF patients, it offered a safe and efficacious alternative to standard parenteral therapy for acute pulmonary exacerbations, despite its possible side effects, such as cartilage toxicity, sunlight sensitivity rash [28] and rapid emergence of resistance [3]. Levofloxacin is a third-generation fluoroquinolone with Gram-positive and Gram-negative antibacterial activity. This fluoroquinolone is used for oral and intravenous administration, and other formulations are being investigated [57].
-
Beta-lactams
Beta-lactams that are used to fight pseudomonal infections in CF patients include aztreonam, anti-pseudomonal carbapenems (doripenem, imipenem–cilastatin and meropenem), penicillins (piperacillin–tazobactam and ticarcillin–clavulanate) and cephalosporins (ceftazidime and cefepime) [58, 59]. In contrast to aminoglycosides and similarly to fluoroquinolones, beta-lactams do not have their antibacterial activity impaired in sputum from CF patients [14, 16]. Aztreonam, active against aerobic Gram-negative bacteria, is available as both intravenous (for acute pulmonary exacerbations) and aerosolised (against chronic airway P. aeruginosa infection) formulations. Carbapenems, active against anaerobic, aerobic Gram-positive and Gram-negative bacteria, are administered by intravenous route in the treatment of acute pulmonary exacerbations [59]. Among cephalosporins, generally active against both aerobic Gram-positive and Gram-negative bacteria, only ceftazidime and cefepime have activity against P. aeruginosa. They are often used to treat P. aeruginosa-related exacerbations by intravenous administration. The penicillins indicated above also have a wide array of antibacterial activity against anaerobic, aerobic Gram-positive and Gram-negative bacteria, including P. aeruginosa, and are administered by the intravenous route in acute pulmonary exacerbations [60].
-
Colistin
Colistin, or polymyxin E, first marketed in the 1950s, has been neglected up until very recently because of reports of nephrotoxicity and neurotoxicity, but multi-resistance to other antibiotics brought it back on stage as a last-resort anti-infective drug and promoted modern pharmacokinetic investigations and re-evaluation of dosing schemes [61]. Colistin is commercially available as colistin sulphate for topic use (oral tablets) and as an inactive prodrug, colistin methanesulphonate, or colistimethate, sodium salt, which is given by intravenous route or via inhalation, alone or in combination therapy to eradicate early infections and to stabilise chronic infections [62]. Colistin sulphate is not used for inhaled therapy due to throat irritation, cough and severe bronchoconstriction. Well tolerated by inhalation, colistimethate sodium is currently used to treat chronic endobronchial P. aeruginosa infection as an extemporaneously prepared solution for inhalation [63, 64].
-
Miscellaneous
Some other antibiotics may be used in specific situations for the treatment of CF lung disease, not only against P. aeruginosa but also against S. maltophilia, S. aureus and other bacterium.
Fosfomycin is an antimicrobial agent with activity against Gram-positive and Gram-negative bacteria, including Pseudomonas spp. and, more specifically, multidrug-resistant Pseudomonas [65]. This antibacterial agent is available in oral formulations as fosfomycin calcium or fosfomycin trometamol, and in intravenous formulation as fosfomycin disodium. The repeated use of the same antibiotics usually leads to the development of resistance, patient intolerance and side effects. In these patients, fosfomycin may be co-administered with other antibiotics [66] because its action mode is considered as unique (inhibition of the initial step in cell wall synthesis) [19] and not affected by other classes of antibiotics, preventing cross-resistance [65]. Mirakhur et al. noticed that intravenous fosfomycin in combination with other antibiotics for pulmonary exacerbations in CF patients colonised by multi-resistant P. aeruginosa resulted in clinical improvement with low side effects [66].
Azithromycin therapy effectively improves lung function and reduces the frequency of pulmonary exacerbations in CF patients chronically colonised with P. aeruginosa, but these effects appear to be only temporary. Some authors hypothesised that the mechanism of azithromycin therapy failure could be linked to its antimicrobial properties and the development of resistance in organisms resident in the CF airway microbiome. Studies have shown that the long-term exposure of CF patients to azithromycin leads to significantly increased macrolide resistance in CF pathogens, such as Haemophilus spp. [22].
Minocycline (by oral administration) may have an “adjunct role”, alone or in combination with other antibiotics in the antimicrobial therapy of multidrug-resistant, respiratory pathogens in CF [19]. More specifically, Kurlandsky and Fader reported that minocycline had in vitro activity against isolates of B. cepacia, S. maltophilia and, to a lesser extent, P. aeruginosa cultured from the respiratory tract of patients with CF lung disease [67].
Clindamycin and rifampicin are recommended for therapy against S. aureus by oral or intravenous route [23]. Linezolid, an oxazolidinone antibiotic, is an optional treatment against MRSA infections in CF patients (with acute exacerbations) [68]. Due to its unique mechanism of action, the probability of cross-resistance with other antibacterial agents is low [69]. In addition, linezolid has a good tissue penetration (especially in the respiratory tract), which is an advantage in the treatment of CF lung disease [70]. However, this antibiotic is expensive and clinical experience is limited [23]. The combination of amoxicillin with clavulanic acid by oral administration may also be used when both S. aureus and H. influenzae infections are present [23].
Other therapeutic agents
-
Gene therapy
Gene therapy is under investigation and investigational approaches are focused on molecules that restore CFTR function or structure [4, 71, 72]. Although the lung epithelium may be easy to reach by inhaled gene vectors as a targeted treatment, efficient and safe formulations of viral and non-viral vectors are still awaited [4, 73].
-
Antimicrobial peptides
Antimicrobial peptides such as lysozyme and lactoferrin are present in the ASL and their antimicrobial [2] (and anti-inflammatory) activity have been investigated because the levels of these peptides are increased in CF patients. However, the high salt concentration resulting from the CFTR dysfunction reduces their activity [19, 74, 75]. Nevertheless, the innate airway defence offers the opportunity for the development of novel therapeutic approaches [75]. Secretary leukocyte proteinase inhibitor and pre-elafin are other resident lung molecules that demonstrated antimicrobial activity against P. aeruginosa. The aim now is to determine the role of protease inhibition in CF lung disease [19, 75, 76].
-
Vaccination
The prevention of P. aeruginosa infection has an important role in the treatment of CF patients. Some authors concluded that anti-pseudomonal vaccination could be effective in preventing P. aeruginosa lung infection [35, 36, 77], and some concluded that nasal and oral vaccines are promising candidates for inducing a specific antibody response in the lungs of CF patients [35, 36].
Indications of anti-pseudomonal inhaled antibiotics according to CF disease evolution
Antibiotic resistance and more pathogens in this disease lead to new strategies in the life-long treatment of pulmonary infection [19]. Some authors described the airways as the major therapeutic target in this disease. Antibiotics given systemically enter the bronchial secretions from blood by simple passive diffusion and some of them diffuse poorly across lipid membranes and into bronchial secretions [3]. Therefore, some drugs are delivered via inhalation to allow high doses of drug at the site of action and decreasing systemic absorption and side effects [4, 16, 19]. Thus, inhalational drug formulations are an attractive and interesting mode of delivery of some drugs to treat CF patients [4] and, more particularly, inhaled antibiotics may be used as prophylaxis, to eradicate early infection, to suppress chronic infection or to treat acute pulmonary exacerbations [16].
Prophylaxis
Regarding prophylactic therapy, paucity of data supports its use and it also has some risks, such as cumulative drug toxicity and the investment of money and time in something unproven [6, 16], but CF patients cannot recuperate previous pulmonary function after pulmonary exacerbations and, therefore, aerosolised antibiotics may control this problem [14] .
Early eradication
Until now, there are no convincing data showing that the eradication of P. aeruginosa improves the long-term prognosis [6, 16]. However, an eradication protocol for the first appearance of P. aeruginosa has been developed in CF patients between 1995 and 2009 and included 2 weeks of intravenous piperacillin and tobramycin, followed by oral ciprofloxacin for 3 weeks and nebulised colistimethate for 6 months. The results showed clinical, economic and resource utilisation benefits [78]. Other studies have been performed, such as inhaled tobramycin alone [7, 79], tobramycin with oral ciprofloxacin [7] and nebulised colistin with oral ciprofloxacin [6, 7, 80], and all of them showed good efficacy. Nevertheless, there is a lack of evidence from large randomised trials to define the optimal drug(s), doses, delivery methods and duration of treatment for early P. aeruginosa eradication [16]. Although more approaches have been described, there are mainly two prospective multi-centre studies (ELITE in Europe, EPIC in USA) that tried to determine the best treatment regime with less adverse effects [6, 80]. EPIC compared different regime therapies: participants inhaled tobramycin with either oral ciprofloxacin or placebo during treatment cycles, but the results showed no significant differences between them [1, 81, 82]. ELITE concluded that TOBI® twice daily for 28 days was as effective as 56 days and was a well-tolerated therapy for early P. aeruginosa infection in CF patients [17]. In this study, almost all of the randomised patients had negative cultures for P. aeruginosa one month after the end of treatment and the majority continued to have negative cultures 27 months later, but clinical outcomes were not compared between patients who did or did not become culture-negative for this bacterium. Nevertheless, a recent 3-year prospective cohort study of adult patients with CF from Canada examined the clearance of P. aeruginosa from their sputum and its relationship to the clinical status of patients. The study concluded that changes in the P. aeruginosa sputum culture status may not reliably predict an improvement in clinical status [83].
Chronic infection treatment
The best evidence regarding the indication of antibiotic aerosol is chronic infection with P. aeruginosa [6]. According to the Cystic Fibrosis Foundation, inhaled tobramycin was recommended for chronic use to improve lung function and reduce pulmonary exacerbations (for children less than 6 years old, no recommendation was made) [16, 49], but the new European guidelines recommended inhaled antibiotics for CF patients irrespective of lung function [6, 16, 49]. Inhaled colistimethate is also used in Europe against chronic infection but there were little evidence about its benefit [6].
Treatment of pulmonary exacerbations
These exacerbations in CF are usually treated with oral or, more frequently, intravenous antibiotics and, as patients recover, inhaled antibiotics are also used to suppress growth of the bacterium [4, 49]. Some authors demonstrated that inhaled antibiotics reduce both the frequency of pulmonary exacerbations and the number of hospitalisations [14]. However, due to the risk of toxicity, the Cystic Fibrosis Foundation concluded that “there is insufficient evidence to recommend for or against continued use of inhaled antibiotics in patients treated with the same antibiotics intravenously for the treatment of an acute exacerbation” [3, 49] and, thus, further studies are necessary [16]. Nevertheless, both chronic and airway clearance therapies may be continued during an exacerbation [49]. Different antibiotics are administered according to colonised pathogens in CF patients. As previously described, P. aeruginosa was usually eradicated with at least two antibiotics, including intravenous aminoglycosides, intravenous and oral fluoroquinolones, and intravenous beta-lactams [4]. According to the Cystic Fibrosis Foundation, “once-daily dosing of aminoglycosides is preferable to 3-times daily dosing” [49], as reported for tobramycin [17]. In order to eradicate other pathogens, other antibiotics such as amoxicillin–clavulanic acid, clindamycin, linezolid co-trimoxazole and macrolides were also used [3, 4, 84].
New perspectives of treatment by inhalation in CF patients
Lung particularities
In order to understand the advantages due to inhaled antibiotics in the treatment of infection by P. aeruginosa in CF lung disease, some issues related to lung characteristics have been taken into account and are described below.
Briefly, the lungs may be divided into two main zones: respiratory and conducting zones [85]. The main defence mechanism of the conducting zone (trachea, bronchi and terminal bronchioles) is the mucociliary escalator and polymorphonuclear neutrophils (PMNs) recruited from the respiratory zone. This zone has cilia, goblet cells and submucosal glands. In the respiratory zone (respiratory bronchioles and alveoli), the defence system consists of alveolar macrophages and defensins. In respect of P. aeruginosa, the non-mucoid phenotype is located mainly in the conductive zone, while mucoid strains are located in both respiratory and conductive zones [1].
It should be noted that pulmonary delivery is a route of administration that may be used as a means of systemic delivery, an alternative to parenteral routes, when the drug has poor bioavailability by the oral route of administration [86]. Some lung features, such as the large surface area for absorption, the high solute permeabilities and the limited proteolytic activity, make this method preferable to administering some drugs (e.g. α2-adrenoceptor agonists, corticosteroids, antimycobacterials and therapeutic macromolecules such as insulin) [87]. As reported by Patton, in order to be absorbed from the lung into the blood, a macromolecule must pass through several barriers, including the monolayer of surfactant, the surface lining fluid (airway fluid contains mucus in contrast to alveolar fluid), the epithelium, the interstitium and the basement membrane and the vascular endothelium [88].
The focus of the present review is on inhalation therapy as a means to provide high local doses and limited systemic exposure [86, 89, 90]. As reported by Pilcer and Amighi, local delivery of drugs to the lungs is desirable, particularly in patients with specific pulmonary diseases such as CF, asthma or chronic pulmonary infections. The principal advantages include reduced systemic side effects and higher drug doses at the site of drug action [91].
A study in the Copenhagen Cystic Fibrosis Centre concluded that the intensive antibiotic therapy used at that institution for chronic P. aeruginosa infections appears to restrain but not eradicate the bacteria from the conductive zone, while the remaining healthy respiratory zone may be protected, for a prolonged period, from massive biofilm infection. This strongly suggests that the conductive zone serves as a bacterial reservoir where the bacteria, organised in mucoid biofilms within the mucus, are protected against antibiotics and host defences [92]. Other authors concluded that nebulised colistin and tobramycin [93] reached high concentrations in the conductive zone (sputum), but the concentrations in the respiratory zone were very low. On the contrary, antibiotics given via oral or intravenous routes reached very low concentrations in the sputum and high concentrations in the respiratory zone due to direct transport by the alveolar blood capillaries [1]. Thus, it is easy to understand the importance of combined oral, intravenous and inhaled therapy to achieve high drug concentrations throughout the respiratory tract.
The importance of particle deposition in the respiratory tract
Understanding the process of particle deposition in the respiratory tract is of great value to improve efficiency in the drug delivery of inhaled drugs [94]. The main forces that are involved in the deposition of particles include gravity, inertia and impulse transfer from collisions with gas molecules [95]. Therefore, particles are deposited by three main basic mechanisms, including sedimentation, inertial impaction and diffusion toward the surfaces of the respiratory tract [94, 95], as represented in Fig. 3.

Schematic diagram representing particle deposition in the lungs according to three main mechanisms related to particle size. It is evident that the smaller particles are deposited in the lower airways as opposed to the bigger particles. Reproduced from reference [94], with permission
Gravitational sedimentation and inertial impaction are the main mechanisms of deposition of particles over 0.5 μm and below 5 μm aerodynamic diameter (da) in the small conducting airways [91]. Impaction occurs principally at the airway bifurcations. It is the main deposition mechanism in extra-thoracic and large conducting airways and for particles with particle size >5 μm. Diffusion, which results from a Brownian motion caused by the impact of surrounding air molecules, is the main deposition mechanism in small airways and alveoli, and most common for particles <0.5 μm [85, 91].
The aerodynamic diameter is defined as the diameter of a sphere with a unit density (ρ) that has the same terminal settling velocity in still air as the particle in consideration [91, 94, 96]. For efficient lung deposition, inhalation aerosols should have aerodynamic diameter in the 1 (or 0.5) to 5 μm range [1, 91, 97]. Larger particles (>5 μm) impact in the upper airway and smaller particles (<1 μm) are easily exhaled [1, 97]. If particles have a density close to one, aerodynamic diameters and geometric diameters (dg) are the same, which is the case in most of the commercialised forms (solid powder or nebulised liquids). However, there might be some advantages of using low-density particles of larger geometric diameter (given that \( {{\mathrm{d}}_{\mathrm{a}}}={{\mathrm{d}}_{\mathrm{g}}}*\sqrt{\rho } \)), while keeping the aerodynamic diameter within the optimal range [91]: easier powder dispersion and lower macrophage uptake. Thus, the key to the success of the controlled-release particles might be the highly porous nature with low density, i.e. ρ ∼ 0.4 g/cm3 [98].
The aerodynamic size of particles can be determined in vitro with cascade impactors. The mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD) are generally reported in the particle characterisation in order to estimate the particle deposition in lungs. Several mathematical models are used in predicting the deposition patterns of particles in the lung, even considering different disease states.
Additionally, there are two other mechanisms of particle deposition, known as interception and electrostatic precipitation, which are related to particle shape and electrostatic charges, respectively [94], but are not discussed herein.
Particle deposition is influenced by other factors such as the pulmonary physiology of patients and different pulmonary diseases, including CF lung disease [91, 94]. In addition, patient factors can affect the efficiency of inhaled therapy, particularly the respiration rate and tidal volumes. Optimal conditions include slow and rhythmic inspiration in order to promote a more homogeneous distribution of drug particles [14].
Additionally, some issues complicate the aerosol formulation, such as drug loss during inhalation, dosing difficulties, enzymatic degradation within the lung and high costs of production. To overcome these, a formulation must be able to be incorporated into an aerosol form and to remain stable against forces generated during aerosolisation. In addition, it should target a specific site or cell population in the lung, protect the compound against aggressive elements in the pulmonary tract and release the compound in a pre-determined mode within an acceptable period of time without producing toxic byproducts. It should also be inert to the surrounding tissue and contain no irritating or toxic additives [99].
CF sputum: a barrier to pulmonary administration
Drug delivery to the deep lung may be impeded by changes such as mucus hypersecretion or thickening, fibrosis or poor blood circulation. Thus, a deep understanding about the impact of the disease (in this case, CF lung disease) on lung pathophysiology is required in order to avoid or reduce the risk of failing to deliver an inhaled particle [86]. More particularly, Ibrahim et al. described the CF sputum as a physical, chemical and biological barrier and as a stage for bacterial resistance [4]. Normal mucus consists of a high percentage of water (90–95 %) and a small amount of mucins, DNA, lipids, electrolytes, proteins and cells. This mucus is easily traversed by gas, ions, nutrients and proteins, and can protect the organism against foreign substances such as toxins and pathogens [4, 100]. CF sputum has less water (90 %) and intact mucins, and more DNA and actin, which provides a higher viscosity to mucus [4]. In addition, mucins have negative charge (carboxyl groups) and form disulphide bonds, physical entanglement and non-covalent interactions [4, 101]. P. aeruginosa is able to evade antibiotic therapies, changing into mucoid strains and forming biofilms. These biofilms, as mentioned above, are resistant to phagocytosis and antibiotics. Their content in oxygen and nutrients are low, which slows down the growth of the bacteria there and reduce their susceptibility to some antibiotics.
Sputum has to be traversed by some drugs, including gene therapeutics and ion-channel regulators. Other drugs such as aerosolised antibiotics have to penetrate the sputum, where they should be evenly distributed [4]. Thus, it is very important for drug delivery to understand the interaction between the drug and CF sputum. Some interactions between sputum and drugs are reported, such as tobramycin, which provides electrostatic interactions with mucus and biofilms, leading to impaired activity, as previously discussed in this review [19].
In addition, DNA and actin have the ability to form a polymer that increases the sputum viscosity [4, 102] and, for example, nanoparticles (e.g. used for the delivery of genetic therapeutics) have difficulty in moving through the viscous sputum.
Some authors proposed modifying the nanoparticles’ surface with polymers such as polyethylene glycol (PEG) with low molecular mass to reduce the interactions between these particles and sputum, to limit their aggregation and to decrease alveolar clearance [4, 103, 104]. The administration of mucolytics before nanoparticles administration may reduce the steric hindrance of mucus, but have two types of outcomes [4]: (1) the delivery of particles with deoxyribonuclease (DNase) and adenoviral genes is enhanced by mucolytics such as N-acetylcysteine [4, 105–108]; (2) gene transfection efficiency is, however, not improved [4, 108]. Pulmonary co-administration of antibiotics and mucus-thinning agents may also be a good tool for local therapy in CF patients. As reported by some authors, an inhalable dry-powder system co-delivering DNase and ciprofloxacin may kill the bacteria in sputum more efficiently than particles with ciprofloxacin alone [4, 109]. Some authors proposed the osmotic agent mannitol as an alternative to DNase and observed that it improved the antibacterial efficiency of ciprofloxacin against P. aeruginosa. Mannitol has the ability to increase the local water content in the mucus and enhances drug penetration into it [110, 111].
Antibiotic aerosol solutions
Aerosolised antibiotics are generally well tolerated, but some adverse effects have been reported, such as cough and throat irritation, which are more frequently observed with dry-powder formulations. Dysgeusia and a decrease in pulmonary function after administration have also been reported. Nevertheless, these effects appear to be more related to aerosolised particles than to the direct pharmacological effects of the drug [14].
In addition, nebulised drugs must be more potent and the time of administration should be reduced in order to improve compliance to therapy. The concomitant administration of nebulised antibiotics may form precipitates and may lead to antibiotic resistance owing to antibiotic dispersion into the ambient air [46]. Therefore, dry-powder formulations may avoid these problems, as will be discussed herein.
As described previously, thickened mucous secretions result in areas of low oxygen concentrations, which lead to reduced potency/activity of aminoglycosides and beta-lactams/monobactams, but less so for fluoroquinolones agents. These differences may have important therapeutic implications [2]. As reported by Bolon, fluoroquinolones result from the addition of a fluorine atom at position 6 of quinolones, a chemical alteration that improves both drug penetration into the bacterial cell and activity against Gram-negative bacteria [43]. With respect to this class of antibiotics, levofloxacin (inhaled) is being developed as a solution for use in the eFlow mesh nebuliser. A recent study evaluated the pharmacokinetics and safety of levofloxacin (single 180-mg dose of two formulations, followed by a multiple-dose phase of 240 mg) and showed high sputum and low serum levofloxacin concentrations [16, 112]. Additionally, Cmax/MIC and AUC/MIC ratios in the airways were substantially larger than with oral or intravenous administration. A phase 2b trial with levofloxacin (inhaled) demonstrated a reduction of P. aeruginosa in sputum and improvements in lung function. The phase 3 trial has been completed (Fig. 4). According to previous information, levofloxacin appears to be more potent against P. aeruginosa biofilms than aminoglycosides and aztreonam. Thus, a high level of levofloxacin readily achievable in the lung following aerosol delivery might be useful for the management of pulmonary infections in CF patients [17].

The Cystic Fibrosis Foundation dynamic ‘pipeline’ of anti-infective therapy. Reproduced from the Cystic Fibrosis Foundation [151], with permission
With respect to beta-lactams, aztreonam, which has antibacterial activity against Gram-negative aerobic bacteria, has been proposed as an aerosolised formulation to treat CF patients, although intravenous aztreonam formulation appeared to be not appropriate for pulmonary administration because it contains arginine, leading to airway inflammation [19]. To overcome this problem, the drug has been reformulated as a lysine salt for inhalation [16] and showed improvements in pulmonary function and reduction in bacterial density in the lungs of CF patients with chronic airway Pseudomonas infection [1, 113, 114]. Aztreonam lysine for inhalation solution (AZLI; Cayston®; Gilead Sciences, Foster City, CA, USA) has recently been developed as an inhaled antibiotic and was approved by the US Food and Drug Administration (FDA) in 2010 for the treatment of chronic P. aeruginosa infection in patients with CF (Fig. 4). This formulation is administered with the Altera® Nebulizer System, which uses eFlow® electronic nebuliser technology with a vibrating, perforated membrane that can generate particles with optimal particle size for delivery to the small conducting airways and over a period of about 2–3 min (highly efficient delivery compared with jet nebulisers) [114–116].
Tobramycin (TOBI® Novartis Pharmaceuticals, Basel, Switzerland) is currently available as a nebulised solution (Fig. 4), as previously described [4]. Recently, fosfomycin in combination with tobramycin [93] has been investigated for inhalation in a phase 2 study, with promising activity against P. aeruginosa [1, 16, 117].
Dry-powder formulations
These formulations are particularly useful for potent drugs, but they have also been used for less potent drugs, such as antibiotics, leading to the administration of higher doses, which reduce patient tolerance. It may be necessary to administer drugs for several minutes and multiple doses when higher doses are required [14]. However, dry-powder formulations are more time-efficient and more hygienic than nebulised liquid formulations [4]. As reported by Høiby, dry-powder inhalers may deliver each dose in one-third of the time required for nebulisation [1]. Being able to deliver anti-pseudomonal therapeutics with a portable inhaler in a fraction of the time required to deliver the same drug via nebulisation is expected to improve quality of life and compliance by CF patients [118].
Currently, inhalable dry powders of some anti-infective drugs, including ciprofloxacin, gentamicin, tobramycin and colistin, have been studied in clinical trials [4].
Inhalable dry powder is being developed for ciprofloxacin, alone or in association with recombinant DNase, which may be a promising strategy for local anti-pseudomonal therapy [17]. Additionally, inhaled ciprofloxacin hydrochloride [log P (o/w) = 0.28] is rapidly absorbed into the systemic circulation, with peak serum levels observed within a few minutes post-inhalation. A dry-powder, controlled-release formulation of ciprofloxacin betaine as a micronised drug coated with a porous layer of phospholipid is currently in clinical development for the treatment of chronic P. aeruginosa infections in CF patients. Insoluble betaine salt at neutral pH dramatically increases the lung residence time [118]. Taking into account that co-encapsulating two or more antibiotics reduces the amount of powder to be inhaled, particularly if the drugs are synergistic [46], Adi et al. evaluated formulations containing ciprofloxacin, doxycycline, or a combination of both, with polyvinyl alcohol (PVA) in order to obtain a controlled-release pattern. The results demonstrated good particle size distribution, thermal stability, acceptable aerosol performance and modified release profiles [119]. As reported by Tsifansky et al., the development of antibiotic resistance in CF patients may be avoided by the concomitant administration of antibiotics in the same formulation, such as microparticles with ciprofloxacin and ceftazidime with dipalmitoyl phosphatidylcholine (DPPC), albumin and lactose as excipients and prepared by the spray-drying method [46]. In addition, the combination of co-spray-dried mannitol and ciprofloxacin from a dry-powder inhaler appears to be an attractive approach to promote mucus clearance in the respiratory tract while simultaneously treating local chronic infection in CF patients. In this case, the delivery of both an osmotic agent (such as mannitol) and an antibiotic in one single dry-powder dose could have many advantages, such as physical stability and an efficient aerosol powder, but further studies are required in order to understand the in vivo antibacterial and mucociliary clearance enhancement of these formulations [111].
With respect to aminoglycosides, tobramycin inhalation powder (TIP), a novel dry-powder formulation (total dose 112 mg), may allow a faster and more convenient dosing regimen compared to the tedious inhalation of nebulised solutions [16, 17]. TIP produced comparable systemic exposure to 300 mg tobramycin solution for inhalation, in less than one-third of the administration time but with cough and dysgeusia as the most common adverse events [17], and the phase 3 trial has been completed (Fig. 4).
Colistin has also been formulated as colistin sulphomethate (i.e. methanesulphonate) dry-powder inhaler (Twincer® inhaler) and a clinical study showed that it was well tolerated by CF patients. However, optimisation regarding particle size and internal resistance of the inhaler is necessary to achieve equivalent pulmonary deposition to liquid nebulisation [120]. A dry powder for inhalation (Colobreathe®) has recently received European marketing approval. Both colistimethate solution and dry powder for inhalation were found to be as effective against P. aeruginosa in chronically infected CF patients. Contrary to inhaled tobramycin solution, inhaled colistimethate solution did not improve lung function [121]. Colistimethate dry powder for inhalation was, however, found to be non-inferior to inhaled tobramycin solution towards lung function preservation, and is more convenient in terms of administration procedure (1 min twice a day vs. 20 min twice a day) and inhaler maintenance [122].
In spite of the advantages reported for dry-powder formulations, i.e. mainly a shorter duration of administration required over a solution of inhalation, there are some issues that need improvement. The deposition of high amounts of dry powders on pulmonary epithelia may lead to adverse effects, ranging from unpleasant taste, cough, throat irritation, dysphonia to haemoptysis, difficulty of breathing and bronchospasm or even acute toxicity [64, 123]. In addition, drug powders are immediate-release dosage forms that lead to fluctuations of drug concentrations and necessitate frequent administrations. To minimise these problems, inhalable innovative drug delivery systems have been under investigation [123] and will be discussed in the following sections.
Innovative aerosol formulations
Sustained release of drugs in the lung may be achieved with their incorporation in liposomes and other formulations, such as biodegradable microspheres [89]. Controlled-release formulations may increase and sustain lung concentration, which contribute to better patient compliance due to a decrease of dosing frequency and may reduce systemic and/or local toxicity. Owing to these characteristics, these formulations appear to be a good “tool” to deliver some anti-infective drugs to treat P. aeruginosa lung infection in CF patients.
However, the development of controlled-release formulations for the lungs may be complicated by the multiple clearance pathways (e.g. mucociliary clearance, macrophages clearance, systemic absorption, cough clearance) and by concerns related to the safety and slow clearance of retentive excipients (e.g. polymers) [118].
Liposomal formulations
Liposomes are one of the most investigated systems for controlled pulmonary delivery, since they may be prepared with phospholipids endogenous to the lung as surfactants and may also incorporate both hydrophilic and hydrophobic drugs [89]. Liposomes release the drug over time and might reduce the dose frequency to once a day or less [16], which may be important for CF patients’ compliance. These formulations appear to be able to maintain their integrity following nebulisation, can penetrate mucoid biofilms [17, 118] and prolong the residence time of the antibiotics in the lungs [16]. Triggering of antibiotic release from liposomes is mediated by the rhamnolipids-produced biofilm-localised bacteria [4]. However, other authors consider that the nebulisation of liposomal formulations can cause structural disruption with consequent release of encapsulated drug, and these formulations are unstable during storage (even at low temperature). Thus, dry liposome powder for inhalation may improve stability, which represents an advantage of these forms [124].
Tobramycin liposome has shown significant increases in both drug retention in the lung and antimicrobial activity compared with classical formulations. However, the clinical demonstration of the sustained release and long-term efficacy could not be demonstrated [19].
Sustained-release lipid formulation of amikacin (Arikace™) is being developed for inhalation. It is delivered in 10–13 min by Pari eFlow and showed superior clinical benefit (e.g. improved lung function) and prolonged time intervals between exacerbations as compared to placebo [16, 17]. In a phase 2 study, Arikace™ administered once daily for 28 days was well tolerated [17] and a phase 3 study is being developed, as demonstrated in Fig. 4. The enzymes in sputum and factors associated with P. aeruginosa can help amikacin release from the liposomes, thus, targeting the drug to the bacterial microenvironment.
Some authors developed liposomal gentamicin formulation to compare its activity with free gentamicin against P. aeruginosa. The results showed that the liposomal formulation protects the drug from bacterial enzymes and facilitates its diffusion across the bacterial envelope, and has an MIC significantly lower than that of corresponding free gentamicin [125, 126].
Also, a liposomal ciprofloxacin formulation for nebulisation is under development. A study with CF patients treated with liposomal ciprofloxacin showed both a decrease in sputum density and an increase in lung function [16]. The hydrochloride salt of ciprofloxacin described in dry-powder formulations has also been encapsulated in liposomes [118].
Microspheres as a new approach for the treatment of CF patients
Microspheres may be an alternative to deliver some of the therapeutic agents previously described. These formulations may facilitate the dispersion of drugs in the inhaled air and reduce the deposition in the oropharynx through adequately modulated physical properties, and may target specific regions of the respiratory tract by modifying the size and density ratios of the particles [95]. Additionally, it is possible to control the kinetics using adequate polymers and formulation additives. This innovative formulation may reduce the direct contact of highly concentrated drug formulations with the lung tissue (and reduce the toxicity) and allows a sustained release [123]. Therefore, microspheres are expected to overcome the problems related to dry-powder inhalers and nebulised solutions of anti-infective drugs for the treatment of CF patients. In addition, microspheres appear to be more stable, from the physicochemical point of view, in comparison to liposomes and, thus, a slower release rate and a longer duration of action may be obtained. Thus, biodegradable microspheres may be more easily formulated in a suitable pulmonary delivery in comparison with liposomes due to higher stability [89, 91]. Microspheres (polymeric or non-polymeric) may be developed to achieve drug targeting and to improve drug absorption, and, at the same time, with fewer adverse effects and lower dosing frequency, maintaining the therapeutic effect [87].
Currently, several inhaled microspheres for lung diseases such as asthma and chronic obstructive pulmonary disease (COPD), pulmonary arterial hypertension, tuberculosis and lung cancer are under investigational research and appear to be a good alternative to lung delivery, as presented in Table 2. Thus, we can understand how these controlled-release formulations may represent an interesting mode of drug delivery in CF patients.
Beyond the scope of the present review, issues related to these formulations are discussed elsewhere and include the methods by which they are formulated: spray-drying technology [4, 127], interfacial polymerisation [128], emulsion solvent evaporation [128], membrane emulsification [129] or coacervation [130]. Regardless of the method, they should have some common characteristics, such as ease of scaling up, accurate and reproducible control over the size, uniformity of the particles (particularly important in lung administration) and compatibility with drug (high temperature, organic solvents or physical forces may affect the bioactivity of the drug) [128].
Applications of polymeric microspheres
Microspheres may be composed of a biodegradable polymer matrix in which the drug is distributed, presenting some advantages, such as the possibility of encapsulation of many types of drugs, general biocompatibility (low immunogenicity and toxicity [98]) and high bioavailability, and ability of sustained release for long periods of time. Drug release may be affected by some factors, including the type of polymer, polymer molecular weight, copolymer composition, nature of excipients and microsphere size [128].
These polymers may be used to develop microspheres for pulmonary delivery as a formulation that provides a sustained drug release and allows high local drug concentration and fewer adverse effects. This scenario leads to fewer daily administrations, and, therefore, better patient compliance to the therapy, which may be important in CF patients.
-
Poly(D,L-lactic-co-glycolic acid) (PLGA) inhaled microspheres
Particles prepared from PLGA have generated considerable interest in recent years for their use as delivery vehicles for various therapeutic agents. PLGA is, by far, the most common biodegradable polymer that is used for the controlled delivery of drugs due to its early use, well-known biocompatibility [98], biodegradability [123, 127] and safety in biomedical preparations, which has been approved for human use by the FDA [97, 127, 131]. However, its use in the lung is not established yet [97]. Thus, there are some questions about its physiological and immunological toxicity in lungs that should be assessed, as reported by Sakagami and Byron [87]. According to Dailey et al., some polymers such as PLGA may be not suitable for application in the respiratory tract, especially when frequent administration is necessary due to its prolonged degradation rate [99]. Additionally, PLA [poly (lactic acid)] and PLGA showed significant reductions in cell viability compared to lipid particles, and long residence due to slow degradation might lead to pulmonary accumulation of polymers [124]. Nevertheless, Mura et al. have recently concluded that the PLGA nanoparticles may affect the viability of Calu-3 cells, but at concentrations that are too high for clinical use and studies with lung cells (e.g. the A549 cell line) confirmed the low cytotoxicity and the absence of inflammation [132]. Therefore, PLGA appears to be a safe ingredient of microspheres for pulmonary administration in CF patients.
As already mentioned for effective pulmonary drug delivery, the physical properties of the particles are crucial and the particle size range that is optimal for inhalation is 1–5 μm. However, it is also ideal for phagocytosis and particles tend to agglomerate (due to Van der Waals and electrostatic forces) [133] and flow and disperse poorly [87]. For effective pulmonary drug delivery, particles must avoid phagocytosis by alveolar macrophages and should maintain the appropriate aerodynamic diameter during storage. Large porous microparticles may circumvent the previous problems, owing to their high geometric diameters and low density, and smaller aerodynamic diameters improving lung deposition [87, 133], as previously reported in this review paper. Further engineering of the particle surface may improve the dispersion and flow of PLGA microspheres by minimising the particle–particle contact area. In order to optimise the lung delivery of particles, two considerations should be taken into account: size uniformity and mucoadhesion. PLGA and PLA are both poorly adhesive in comparison with other polymers (e.g. polyanhydrides), possibly due to the limited hydrogen bonding potential with mucus glycoproteins. However, they may be coated with polymers with high densities of functional groups (e.g. polyamines) [98] and with polymers such as chitosan, as described below in this review. Regarding PLGA microspheres, they are commonly prepared by the solvent evaporation technique and the water-in-oil-in-water (W/O/W) double emulsion is the most common technique for encapsulating hydrophilic drugs. Emami et al. proposed a single-phase oil-in-oil (O/O) solvent evaporation technique to overcome several difficulties, such as the low entrapment efficiency (EE) due to the leakage of hydrophilic drugs to the outer aqueous phase. This study concluded that a low PLGA concentration and a high stirring speed are necessary to obtain a small mean particle size and a narrow distribution [97].
Yang et al. prepared non-porous PLGA microparticles by a W/O/W double-emulsion method for pulmonary drug delivery. However, most of the dry particles were present in aggregates. Therefore, they aimed to improve the geometric diameter and reduce MMAD to avoid macrophage uptake. Hence, they prepared highly porous large PLGA microparticles employing ammonium bicarbonate (an effervescent porogen which is good for hydrophilic drugs) to create porous microspheres. Subsequently, they encapsulated two model drugs, lysozyme and doxorubicin hydrochloride, in the highly porous large PLGA microparticles, and concluded that this method is an efficient way of making polymeric microparticles for sustained local drug delivery by inhalation. Nevertheless, they tried to encapsulate ciprofloxacin in the highly porous large PLGA microparticles, but the resulting particles showed a very low fine-particle fraction (FPF), because the majority of ciprofloxacin precipitated out as the pH of the internal aqueous phase increased upon the ammonium bicarbonate addition. Therefore, this encapsulation method would be of limited use for the delivery of ciprofloxacin [133].
With respect to tuberculosis and, more specifically, to infection by Mycobacterium tuberculosis, some authors prepared rifampicin-loaded PLGA microspheres. O’Hara and Hickey compared double-emulsion solvent extraction and spray-drying techniques in the production of microspheres for inhalation, taking eight variables for the emulsification in a factorial design and using a Box–Behnken statistical approach to optimise the drug loading and particle size. The authors concluded that the surface of the spray-dried microparticles appeared to have collapsed inwards and a had smaller volume median diameter, in contrast to spherical particles from the emulsion–extraction method [98, 134]. Based on the particle size, aerosol delivery to the lungs can be more effective for the spray-dried products. However, its efficacy has yet to be evaluated in in vivo studies. Recently, Doan et al. have also prepared rifampicin-loaded PLGA microspheres for sustained lung delivery by a dry-in-emulsion [oil-in-water (O/W) or W/O/W emulsions] process with a premix membrane homogenisation step, which was proposed in order to produce monodisperse microspheres with a high productivity [123].
In order to improve the antimicrobial efficacy of antibiotics for tuberculosis treatment, Giovagnoli et al. developed large porous microspheres for capreomycin sulphate pulmonary delivery prepared by the W/O/W double-emulsion solvent evaporation method. The authors concluded that, although this method was poorly applicable to large-scale production, it was easy to implement and preserved PLGA polymer characteristics. The obtained microspheres appeared to be potentially useful as a tool for improving anti-tubercular treatments. More detailed information is given in Table 2. A potential liposomal capreomycin sulphate formulation has also been developed for inhalation use [135].
With respect to pulmonary arterial hypertension, Gupta et al. have recently studied the release of drug over a prolonged period of time and the prevention of enzymatic degradation of prostaglandin E1 by using prostaglandin E1-loaded PLGA-based microspheres, which were formulated by the W/O/W double-emulsion solvent evaporation method. These formulations showed good metabolic stability and produced prolonged release of prostaglandin E1 after pulmonary administration [136].
-
Chitosan inhaled microspheres
In addition to the microspheres formulated with polymers such as PLGA or PLA, natural polymers such as alginate, albumin and chitosan (a natural cationic polysaccharide [127]) have also been used in the preparation of microspheres suitable for pulmonary delivery. They present several advantages, including low cost, compatibility with a wide range of drugs, minimal use of organic solvents, bio/mucoadhesion, stability, safety and approval for human use by the FDA [130]. Bioadhesive properties are related to attractive electrostatic forces between the negatively charged glycoprotein of mucin and the positively charged amino groups of the polymer [137]. These properties allow an efficient absorption and enhanced bioavailability, a much more intimate contact with the mucus layer and a specific targeting of drug to the absorption [138].
Ventura et al. have prepared chitosan microspheres for the intrapulmonary administration of moxifloxacin, a fluoroquinolone that may represent an interesting drug for the treatment of CF lung disease. These microspheres were prepared by the spray-drying method using glutaraldehyde (GL) as the cross-linking agent. The produced microspheres were spherical with suitable sizes for inhalation, as detailed in Table 2. These authors concluded that formulation parameters such as concentrations of chitosan, moxifloxacin and GL affect the microsphere sizes. For example, a higher encapsulation efficiency (EE) was observed for higher chitosan concentrations and the opposite is observed at the highest GL concentrations [137].
As previously described about the strategy of coating polyester polymers with other type of polymers, some authors compared the particle stability during the nebulisation of PLGA, chitosan and chitosan-coated PLGA microparticles in delivering rifampicin to lung macrophages. PLGA microspheres were formulated by an O/W solvent evaporation method, while chitosan microparticles were prepared by the precipitation method and particles with both chitosan and PLGA were prepared by the emulsion solvent diffusion method in water, as described in Table 2. These authors concluded that PLGA polymer is better than chitosan for the preparation of particles that could deliver rifampicin to alveolar macrophages after nebulisation. Nevertheless, a combination of the two polymers leads to the formation of very stable microparticles with high loading capacity for rifampicin, lower cytotoxicity towards alveolar epithelial cells (compared to PLGA microparticles) and equivalent (to chitosan microparticles) mucoadhesive properties [130].
Moreover, several antibacterial (including anti-pseudomonal) microspheres have been prepared for intravenous or ocular delivery, and may represent an interesting tool for the development of inhaled microspheres for CF lung disease, by adjusting some formulation parameters. Some of these formulations are present in Table 2.
Conclusions
Cystic fibrosis (CF) is a severe inherited disease which affects particularly the lungs. Scientific research has been extensively carried out on respiratory infections and their treatment in CF lung disease. These respiratory infections are mainly due to Pseudomonas aeruginosa infection and its treatment includes anti-infective agents. More particularly, combined oral, intravenous and inhaled antibiotics are currently used to treat these airways infections. Combination of antibiotics, including aminoglycosides, beta-lactams and fluoroquinolones, are frequently administered in order to avoid antimicrobial resistance. Moreover, some innovative formulations for inhalation (e.g. liposomes) are under investigation and appear to be a good tool to improve quality of life in CF patients. These formulations can achieve high local concentrations and may reduce antimicrobial resistance. They may also reduce adverse effects and improve patient compliance to therapy. Other controlled-release formulations such as microspheres may represent a new approach in the treatment of this disease.
References
Høiby N (2011) Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 9(1):32–38. doi:10.1186/1741-7015-9-32
Boucher RC (2004) New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23(1):146–158. doi:10.1183/09031936.03.00057003
Döring G, Conway SP, Heijerman HG, Hodson ME, Høiby N, Smyth A, Touw DJ (2000) Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur Respir J 16(4):749–767
Ibrahim BM, Tsifansky MD, Yang Y, Yeo Y (2011) Challenges and advances in the development of inhalable drug formulations for cystic fibrosis lung disease. Expert Opin Drug Deliv 8(4):451–466. doi:10.1517/17425247.2011.561310
Ramsey BW (1996) Management of pulmonary disease in patients with cystic fibrosis. N Engl J Med 335(3):179–188. doi:10.1056/NEJM199607183350307
Girón Moreno RM, Salcedo Posadas A, Mar Gómez-Punter R (2011) Inhaled antibiotic therapy in cystic fibrosis (Antibioterapia inhalada en la fibrosis quistica). Arch Bronconeumol 47(Suppl 6):14–18
Rogers GB, Hoffman LR, Döring G (2011) Novel concepts in evaluating antimicrobial therapy for bacterial lung infections in patients with cystic fibrosis. J Cyst Fibros 10(6):387–400. doi:10.1016/j.jcf.2011.06.014
George AM, Jones PM, Middleton PG (2009) Cystic fibrosis infections: treatment strategies and prospects. FEMS Microbiol Lett 300(2):153–164. doi:10.1111/j.1574-6968.2009.01704.x
Boucher RC (2007) Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58(1):157–170. doi:10.1146/annurev.med.58.071905.105316
Davies JC, Bilton D (2009) Bugs, biofilms, and resistance in cystic fibrosis. Respir Care 54(5):628–640
Mall MA (2008) Role of cilia, mucus, and airway surface liquid in mucociliary dysfunction: lessons from mouse models. J Aerosol Med Pulm Drug Deliv 21(1):13–24. doi:10.1089/jamp.2007.0659
Deneuville E, Perrot-Minot C, Pennaforte F, Roussey M, Zahm JM, Clavel C, Puchelle E, de Bentzmann S (1997) Revisited physicochemical and transport properties of respiratory mucus in genotyped cystic fibrosis patients. Am J Respir Crit Care Med 156(1):166–172
King M (1987) The role of mucus viscoelasticity in cough clearance. Biorheology 24(6):589–597
Dudley MN, Loutit J, Griffith DC (2008) Aerosol antibiotics: considerations in pharmacological and clinical evaluation. Curr Opin Biotechnol 19(6):637–643. doi:10.1016/j.copbio.2008.11.002
Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O’Neal WK, Boucher RC (2003) Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A 100(26):16083–16088. doi:10.1073/pnas.2634339100
Geller DE (2009) Aerosol antibiotics in cystic fibrosis. Respir Care 54(5):658–670
Bals R, Hubert D, Tümmler B (2011) Antibiotic treatment of CF lung disease: from bench to bedside. J Cyst Fibros 10(Suppl 2):S146–S151. doi:10.1016/s1569-1993(11)60019-2
Goss CH, Muhlebach MS (2011) Review: Staphylococcus aureus and MRSA in cystic fibrosis. J Cyst Fibros 10(5):298–306. doi:10.1016/j.jcf.2011.06.002
van Westreenen M, Tiddens HAWM (2010) New antimicrobial strategies in cystic fibrosis. Paediatr Drugs 12(6):343–352
Harrison F (2007) Microbial ecology of the cystic fibrosis lung. Microbiology 153(4):917–923. doi:10.1099/mic.0.2006/004077-0
de Vrankrijker AMM, Wolfs TFW, van der Ent CK (2010) Challenging and emerging pathogens in cystic fibrosis. Paediatr Respir Rev 11(4):246–254. doi:10.1016/j.prrv.2010.07.003
Grinwis ME, Sibley CD, Parkins MD, Eshaghurshan CS, Rabin HR, Surette MG (2010) Macrolide and clindamycin resistance in Streptococcus milleri group isolates from the airways of cystic fibrosis patients. Antimicrob Agents Chemother 54(7):2823–2829. doi:10.1128/aac.01845-09
Döring G, Flume P, Heijerman H, Elborn JS; Consensus Study Group (2012) Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros 11(6):461–479. doi:10.1016/j.jcf.2012.10.004
Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E, Sime-Ngando T, Chabé M, Viscogliosi E (2012) The airway microbiota in cystic fibrosis: a complex fungal and bacterial community—implications for therapeutic management. PLoS One 7(4):e36313. doi:10.1371/journal.pone.0036313
VanDevanter DR, LiPuma JJ (2012) Microbial diversity in the cystic fibrosis airways: where is thy sting? Future Microbiol 7(7):801–803. doi:10.2217/fmb.12.55
Zemanick ET, Sagel SD, Harris JK (2011) The airway microbiome in cystic fibrosis and implications for treatment. Curr Opin Pediatr 23(3):319–324. doi:10.1097/MOP.0b013e32834604f2
Lynch SV, Bruce KD (2013) The cystic fibrosis airway microbiome. Cold Spring Harb Perspect Med. 3(3). pii: a009738. doi:10.1101/cshperspect.a009738
Banerjee D, Stableforth D (2000) The treatment of respiratory Pseudomonas infection in cystic fibrosis: what drug and which way? Drugs 60(5):1053–1064
Rosenfeld M, Ramsey BW, Gibson RL (2003) Pseudomonas acquisition in young patients with cystic fibrosis: pathophysiology, diagnosis, and management. Curr Opin Pulm Med 9(6):492–497
Cramer N, Wiehlmann L, Ciofu O, Tamm S, Høiby N, Tümmler B (2012) Molecular epidemiology of chronic Pseudomonas aeruginosa airway infections in cystic fibrosis. PLoS One 7(11):e50731. doi:10.1371/journal.pone.0050731
Campana S, Taccetti G, Ravenni N, Masi I, Audino S, Sisi B, Repetto T, Döring G, de Martino M (2004) Molecular epidemiology of Pseudomonas aeruginosa, Burkholderia cepacia complex and methicillin-resistant Staphylococcus aureus in a cystic fibrosis center. J Cyst Fibros 3(3):159–163. doi:10.1016/j.jcf.2004.03.010
Lee TWR, Brownlee KG, Conway SP, Denton M, Littlewood JM (2003) Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cystic Fibros 2(1):29–34. doi:10.1016/s1569-1993(02)00141-8
Billard-Pomares T, Herwegh S, Wizla-Derambure N, Turck D, Courcol R, Husson M-O (2011) Application of quantitative PCR to the diagnosis and monitoring of Pseudomonas aeruginosa colonization in 5–18-year-old cystic fibrosis patients. J Med Microbiol 60(2):157–161. doi:10.1099/jmm.0.023838-0
Deschaght P, Van Daele S, De Baets F, Vaneechoutte M (2011) PCR and the detection of Pseudomonas aeruginosa in respiratory samples of CF patients. A literature review. J Cyst Fibros 10(5):293–297. doi:10.1016/j.jcf.2011.05.004
Bumann D, Behre C, Behre K, Herz S, Gewecke B, Gessner JE, von Specht BU, Baumann U (2010) Systemic, nasal and oral live vaccines against Pseudomonas aeruginosa: a clinical trial of immunogenicity in lower airways of human volunteers. Vaccine 28(3):707–713. doi:10.1016/j.vaccine.2009.10.080
Döring G, Meisner C, Stern M; Flagella Vaccine Trial Study Group (2007) A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc Natl Acad Sci USA 104(26):11020–11025. doi:10.1073/pnas.0702403104
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O (2010) Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 35(4):322–332. doi:10.1016/j.ijantimicag.2009.12.011
Nicas TI, Hancock REW (1983) Pseudomonas aeruginosa outer membrane permeability: isolation of a porin protein F-deficient mutant. J Bacteriol 153(1):281–285
Aronoff SC (1988) Outer membrane permeability in Pseudomonas cepacia: diminished porin content in a beta-lactam-resistant mutant and in resistant cystic fibrosis isolates. Antimicrob Agents Chemother 32(11):1636–1639
Burns JL, Hedin LA, Lien DM (1989) Chloramphenicol resistance in Pseudomonas cepacia because of decreased permeability. Antimicrob Agents Chemother 33(2):136–141
Nikaido H (1994) Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264(5157):382–388. doi:10.1126/science.8153625
Grégoire N, Raherison S, Grignon C, Comets E, Marliat M, Ploy M-C, Couet W (2010) Semimechanistic pharmacokinetic–pharmacodynamic model with adaptation development for time–kill experiments of ciprofloxacin against Pseudomonas aeruginosa. Antimicrob Agents Chemother 54(6):2379–2384. doi:10.1128/aac.01478-08
Bolon MK (2011) The newer fluoroquinolones. Med Clin N Am 95(4):793–817. doi:10.1016/j.mcna.2011.03.006
Hocquet D, El Garch F, Vogne C, Plésiat P (2003) Mécanisme de la résistance adaptative de Pseudomonas aeruginosa aux aminosides. Pathol Biol 51(8–9):443–448. doi:10.1016/s0369-8114(03)00167-6
Valenza G, Radike K, Schoen C, Horn S, Oesterlein A, Frosch M, Abele-Horn M, Hebestreit H (2010) Resistance to tobramycin and colistin in isolates of Pseudomonas aeruginosa from chronically colonized patients with cystic fibrosis under antimicrobial treatment. Scand J Infect Dis 42(11–12):885–889. doi:10.3109/00365548.2010.509333
Tsifansky MD, Yeo Y, Evgenov OV, Bellas E, Benjamin J, Kohane DS (2008) Microparticles for inhalational delivery of antipseudomonal antibiotics. AAPS J 10(2):254–260. doi:10.1208/s12248-008-9033-8
Sonnet P, Izard D, Mullié C (2012) Prevalence of efflux-mediated ciprofloxacin and levofloxacin resistance in recent clinical isolates of Pseudomonas aeruginosa and its reversal by the efflux pump inhibitors 1-(1-naphthylmethyl)-piperazine and phenylalanine-arginine-beta-naphthylamide. Int J Antimicrob Agents 39(1):77–80
Govan JRW (2006) Multidrug-resistant pulmonary infection in cystic fibrosis—what does ‘resistant’ mean? J Med Microbiol 55(12):1615–1617. doi:10.1099/jmm.0.46884-0
Flume PA, Mogayzel PJ Jr, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, Marshall BC; Clinical Practice Guidelines for Pulmonary Therapies Committee (2009) Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 180(9):802–808. doi:10.1164/rccm.200812-1845PP
Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O (2008) Tissue concentrations: do we ever learn? J Antimicrob Chemother 61(2):235–237. doi:10.1093/jac/dkm476
Ryan DM, Cars O (1983) A problem in the interpretation of beta-lactam antibiotic levels in tissues. J Antimicrob Chemother 12(3):281–284. doi:10.1093/jac/12.3.281
Kiem S, Schentag JJ (2008) Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother 52(1):24–36. doi:10.1128/aac.00133-06
Conte JE Jr, Golden J, Duncan S, McKenna E, Lin E, Zurlinden E (1996) Single-dose intrapulmonary pharmacokinetics of azithromycin, clarithromycin, ciprofloxacin, and cefuroxime in volunteer subjects. Antimicrob Agents Chemother 40(7):1617–1622
Rose LM, Neale R (2010) Development of the first inhaled antibiotic for the treatment of cystic fibrosis. Sci Transl Med 2(63):63mr4. doi:10.1126/scitranslmed.3001634
Sawicki GS, Signorovitch JE, Zhang J, Latremouille-Viau D, von Wartburg M, Wu EQ, Shi L (2012) Reduced mortality in cystic fibrosis patients treated with tobramycin inhalation solution. Pediatr Pulmonol 47(1):44–52. doi:10.1002/ppul.21521
Ballmann M, Smyth A, Geller DE (2011) Therapeutic approaches to chronic cystic fibrosis respiratory infections with available, emerging aerosolized antibiotics. Respir Med 105:S2–S8
Stockmann C, Sherwin CMT, Zobell JT, Young DC, Waters CD, Spigarelli MG, Ampofo K (2013) Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: III. fluoroquinolones. Pediatr Pulmonol 48(3):211–220. doi:10.1002/ppul.22667
Zobell JT, Young DC, Waters CD, Ampofo K, Cash J, Marshall BC, Olson J, Chatfield BA (2011) A survey of the utilization of anti-pseudomonal beta-lactam therapy in cystic fibrosis patients. Pediatr Pulmonol 46(10):987–990. doi:10.1002/ppul.21467
Zobell JT, Young DC, Waters CD, Stockmann C, Ampofo K, Sherwin CMT, Spigarelli MG (2012) Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: I. aztreonam and carbapenems. Pediatr Pulmonol 47(12):1147–1158. doi:10.1002/ppul.22655
Zobell JT, Waters CD, Young DC, Stockmann C, Ampofo K, Sherwin CMT, Spigarelli MG (2013) Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: II. cephalosporins and penicillins. Pediatr Pulmonol 48(2):107–122. doi:10.1002/ppul.22669
Couet W, Grégoire N, Marchand S, Mimoz O (2012) Colistin pharmacokinetics: the fog is lifting. Clin Microbiol Infect 18(1):30–39. doi:10.1111/j.1469-0691.2011.03667.x
Oliveira MS, Prado GVB, Costa SF, Grinbaum RS, Levin AS (2009) Polymyxin B and colistimethate are comparable as to efficacy and renal toxicity. Diagn Microbiol Infect Dis 65(4):431–434. doi:10.1016/j.diagmicrobio.2009.07.018
Westerman EM, Le Brun PPH, Touw DJ, Frijlink HW, Heijerman HGM (2004) Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J Cystic Fibros 3(1):23–28
Le Brun PPH, de Boer AH, Mannes GPM, de Fraı̂ture DMI, Brimicombe RW, Touw DJ, Vinks AA, Frijlink HW, Heijerman HGM (2002) Dry powder inhalation of antibiotics in cystic fibrosis therapy: part 2: inhalation of a novel colistin dry powder formulation: a feasibility study in healthy volunteers and patients. Eur J Pharm Biopharm 54(1):25–32. doi:10.1016/s0939-6411(02)00044-9
Faruqi S, McCreanor J, Moon T, Meigh R, Morice AH (2008) Fosfomycin for Pseudomonas-related exacerbations of cystic fibrosis. Int J Antimicrob Agents 32(5):461–463. doi:10.1016/j.ijantimicag.2008.05.010
Mirakhur A, Gallagher MJ, Ledson MJ, Hart CA, Walshaw MJ (2003) Fosfomycin therapy for multiresistant Pseudomonas aeruginosa in cystic fibrosis. J Cystic Fibros 2(1):19–24. doi:10.1016/s1569-1993(02)00143-1
Kurlandsky LE, Fader RC (2000) In vitro activity of minocycline against respiratory pathogens from patients with cystic fibrosis. Pediatr Pulmonol 29(3):210–212. doi:10.1002/(sici)1099-0496(200003)29:3<210::aid-ppul9>3.0.co;2–2
Keel RA, Schaeftlein A, Kloft C, Pope JS, Knauft RF, Muhlebach M, Nicolau DP, Kuti JL (2011) Pharmacokinetics of intravenous and oral linezolid in adults with cystic fibrosis. Antimicrob Agents Chemother 55(7):3393–3398. doi:10.1128/aac.01797-10
Dotis J, Iosifidis E, Ioannidou M, Roilides E (2010) Use of linezolid in pediatrics: a critical review. Int J Infect Dis 14(8):e638–e648. doi:10.1016/j.ijid.2009.10.002
Di Paolo A, Malacarne P, Guidotti E, Danesi R, Del Tacca M (2010) Pharmacological issues of linezolid: an updated critical review. Clin Pharmacokinet 49(7):439–447
Grasemann H, Ratjen F (2010) Emerging therapies for cystic fibrosis lung disease. Expert Opin Emerg Drugs 15(4):653–659. doi:10.1517/14728214.2010.517746
Anderson P (2010) Emerging therapies in cystic fibrosis. Ther Adv Respir Dis 4(3):177–185. doi:10.1177/1753465810371107
Boucher RC (1999) Status of gene therapy for cystic fibrosis lung disease. J Clin Invest 103(4):441–445. doi:10.1172/jci6330
Sagel SD, Sontag MK, Accurso FJ (2009) Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr Pulmonol 44(4):402–409. doi:10.1002/ppul.21028
Travis SM, Singh PK, Welsh MJ (2001) Antimicrobial peptides and proteins in the innate defense of the airway surface. Curr Opin Immunol 13(1):89–95. doi:10.1016/s0952-7915(00)00187-4
Albrecht MT, Wang W, Shamova O, Lehrer RI, Schiller NL (2002) Binding of protegrin-1 to Pseudomonas aeruginosa and Burkholderia cepacia. Respir Res 3(1):18
Döring G, Pier GB (2008) Vaccines and immunotherapy against Pseudomonas aeruginosa. Vaccine 26(8):1011–1024. doi:10.1016/j.vaccine.2007.12.007
Lillquist YP, Cho E, Davidson AGF (2011) Economic effects of an eradication protocol for first appearance of Pseudomonas aeruginosa in cystic fibrosis patients: 1995 vs. 2009. J Cyst Fibros 10(3):175–180. doi:10.1016/j.jcf.2011.01.002
Ratjen F, Munck A, Kho P, Angyalosi G; ELITE Study Group (2010) Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax 65(4):286–291. doi:10.1136/thx.2009.121657
Giugno H, Castaños C, Lubatti A, Pinheiro JL, Hernández C, González Pena H (2010) Early antibiotic treatment for eradication of initial infection by Pseudomonas aeruginosa in patients with cystic fibrosis. Arch Argent Pediatr 108(2):141–147
Treggiari MM, Rosenfeld M, Mayer-Hamblett N, Retsch-Bogart G, Gibson RL, Williams J, Emerson J, Kronmal RA, Ramsey BW; EPIC Study Group (2009) Early anti-pseudomonal acquisition in young patients with cystic fibrosis: rationale and design of the EPIC clinical trial and observational study. Contemp Clin Trials 30(3):256–268. doi:10.1016/j.cct.2009.01.003
Mayer-Hamblett N, Burns JL, Khan U, Retsch-Bogart G, Treggiari M, Ramsey BW (2010) Predictors of Pseudomonas aeruginosa recurrence in cystic fibrosis: results from the epic trial. Pediatr Pulmonol 45:326–327
Burkett A, Vandemheen KL, Giesbrecht-Lewis T, Ramotar K, Ferris W, Chan F, Doucette S, Fergusson D, Aaron SD (2012) Persistency of Pseudomonas aeruginosa in sputum cultures and clinical outcomes in adult patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis 31:1603–1610. doi:10.1007/s10096-011-1483-8
Flume PA, O’Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ Jr, Willey-Courand DB, Bujan J, Finder J, Lester M, Quittell L, Rosenblatt R, Vender RL, Hazle L, Sabadosa K, Marshall B; Cystic Fibrosis Foundation, Pulmonary Therapies Committee (2007) Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 176(10):957–969. doi:10.1164/rccm.200705-664OC
Aulton ME (2005) Delineamento de formas farmacêuticas, 2nd edn. Artmed, Porto Alegre
Forbes B, Asgharian B, Dailey LA, Ferguson D, Gerde P, Gumbleton M, Gustavsson L, Hardy C, Hassall D, Jones R, Lock R, Maas J, McGovern T, Pitcairn GR, Somers G, Wolff RK (2010) Challenges in inhaled product development and opportunities for open innovation. Adv Drug Deliv Rev 63(1–2):69–87. doi:10.1016/j.addr.2010.11.004
Sakagami M, Byron PR (2005) Respirable microspheres for inhalation: the potential of manipulating pulmonary disposition for improved therapeutic efficacy. Clin Pharmacokinet 44(3):263–277
Patton JS (1996) Mechanisms of macromolecule absorption by the lungs. Adv Drug Deliv Rev 19(1):3–36. doi:10.1016/0169-409x(95)00113-l
Zeng XM, Martin GP, Marriott C (1995) The controlled delivery of drugs to the lung. Int J Pharm 124(2):149–164. doi:10.1016/0378-5173(95)00104-q
Marchand S, Gobin P, Brillault J, Baptista S, Adier C, Olivier J-C, Mimoz O, Couet W (2010) Aerosol therapy with colistin methanesulfonate: a biopharmaceutical issue illustrated in rats. Antimicrob Agents Chemother 54(9):3702–3707. doi:10.1128/aac.00411-10
Pilcer G, Amighi K (2010) Formulation strategy and use of excipients in pulmonary drug delivery. Int J Pharm 392(1–2):1–19. doi:10.1016/j.ijpharm.2010.03.017
Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Høiby N (2009) Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol 44(6):547–558. doi:10.1002/ppul.21011
Vendrell Relat M, Muñoz Castro G, Sabater Talaverano G, De Gracia Roldán J (2011) The future of inhaled antibiotic therapy. New products (El futuro de la antibioterapia inhalada. Nuevos productos). Arch Bronconeumol 47(Suppl 6):30–32
Carvalho TC, Peters JI, Williams RO 3rd (2011) Influence of particle size on regional lung deposition—what evidence is there? Int J Pharm 406(1–2):1–10. doi:10.1016/j.ijpharm.2010.12.040
Heyder J (2004) Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proc Am Thorac Soc 1(4):315–320. doi:10.1513/pats.200409-046TA
de Boer AH, Gjaltema D, Hagedoorn P, Frijlink HW (2002) Characterization of inhalation aerosols: a critical evaluation of cascade impactor analysis and laser diffraction technique. Int J Pharm 249(1–2):219–231. doi:10.1016/s0378-5173(02)00526-4
Emami J, Hamishehkar H, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H, Mirzadeh H, Fakhari A, Nokhodchi A (2009) Particle size design of PLGA microspheres for potential pulmonary drug delivery using response surface methodology. J Microencapsul 26(1):1–8. doi:10.1080/02652040802083900
Mohamed F, van der Walle CF (2008) Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci 97(1):71–87. doi:10.1002/jps.21082
Dailey LA, Kleemann E, Wittmar M, Gessler T, Schmehl T, Roberts C, Seeger W, Kissel T (2003) Surfactant-free, biodegradable nanoparticles for aerosol therapy based on the branched polyesters, DEAPA-PVAL-g-PLGA. Pharm Res 20(12):2011–2020. doi:10.1023/b:pham.0000008051.94834.10
Cu Y, Saltzman WM (2009) Mathematical modeling of molecular diffusion through mucus. Adv Drug Deliv Rev 61(2):101–114. doi:10.1016/j.addr.2008.09.006
Cone RA (2009) Barrier properties of mucus. Adv Drug Deliv Rev 61(2):75–85. doi:10.1016/j.addr.2008.09.008
Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Fessler MB, Malcolm KC, Vasil ML, Nick JA (2005) Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun 73(6):3693–3701. doi:10.1128/iai.73.6.3693-3701.2005
Suk JS, Lai SK, Wang Y-Y, Ensign LM, Zeitlin PL, Boyle MP, Hanes J (2009) The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials 30(13):2591–2597. doi:10.1016/j.biomaterials.2008.12.076
Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang Y-Y, Cone R, Hanes J (2007) Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A 104(5):1482–1487. doi:10.1073/pnas.0608611104
Sanders NN, De Smedt SC, Van Rompaey E, Simoens P, De Baets F, Demeester J (2000) Cystic fibrosis sputum: a barrier to the transport of nanospheres. Am J Respir Crit Care Med 162(5):1905–1911
Sanders NN, Van Rompaey E, De Smedt SC, Demeester J (2002) On the transport of lipoplexes through cystic fibrosis sputum. Pharm Res 19(4):451–456. doi:10.1023/a:1015139527747
Kushwah R, Oliver JR, Cao H, Hu J (2007) Nacystelyn enhances adenoviral vector-mediated gene delivery to mouse airways. Gene Ther 14(16):1243–1248. doi:10.1038/sj.gt.3302968
Ferrari S, Kitson C, Farley R, Steel R, Marriott C, Parkins DA, Scarpa M, Wainwright B, Evans MJ, Colledge WH, Geddes DM, Alton EW (2001) Mucus altering agents as adjuncts for nonviral gene transfer to airway epithelium. Gene Ther 8(18):1380–1386. doi:10.1038/sj.gt.3301525
Yang Y, Tsifansky MD, Wu C-J, Yang HI, Schmidt G, Yeo Y (2010) Inhalable antibiotic delivery using a dry powder co-delivering recombinant deoxyribonuclease and ciprofloxacin for treatment of cystic fibrosis. Pharm Res 27(1):151–160. doi:10.1007/s11095-009-9991-2
Yang Y, Tsifansky MD, Shin S, Lin Q, Yeo Y (2011) Mannitol-guided delivery of ciprofloxacin in artificial cystic fibrosis mucus model. Biotechnol Bioeng 108(6):1441–1449. doi:10.1002/bit.23046
Adi H, Young PM, Chan H-K, Agus H, Traini D (2010) Co-spray-dried mannitol–ciprofloxacin dry powder inhaler formulation for cystic fibrosis and chronic obstructive pulmonary disease. Eur J Pharm Sci 40(3):239–247. doi:10.1016/j.ejps.2010.03.020
Geller DE, Flume PA, Griffith DC, Morgan E, White D, Loutit JS, Dudley MN (2011) Pharmacokinetics and safety of MP-376 (levofloxacin inhalation solution) in cystic fibrosis subjects. Antimicrob Agents Chemother 55(6):2636–2640. doi:10.1128/aac.01744-10
Assael BM (2011) Aztreonam inhalation solution for suppressive treatment of chronic Pseudomonas aeruginosa lung infection in cystic fibrosis. Expert Rev Anti Infect Ther 9(11):967–973. doi:10.1586/eri.11.131
Plosker GL (2011) Aztreonam lysine for inhalation solution in cystic fibrosis: profile report. Paediatr Drugs 13(2):129–131. doi:10.2165/11206940-000000000-00000
Kirkby S, Novak K, McCoy K (2011) Aztreonam (for inhalation solution) for the treatment of chronic lung infections in patients with cystic fibrosis: an evidence-based review. Core Evid 6:59–66
Plosker GL (2010) Aztreonam lysine for inhalation solution in cystic fibrosis. Drugs 70(14):1843–1855
McColley SA, Trapnell B, Kissner D, McKevitt M, Montgomery B, Rosen J; FTIS Study Group (2010) Fosfomycin/tobramycin for inhalation (FTI): microbiological results of a phase 2 placebo-controlled trial in patients with cystic fibrosis and Pseudomonas aeruginosa. Pediatr Pulmonol 45:338
Weers JG, Bell J, Chan HK, Cipolla D, Dunbar C, Hickey AJ, Smith IJ (2010) Pulmonary formulations: what remains to be done? J Aerosol Med Pulm Drug Deliv 23:S5–S23. doi:10.1089/jamp.2010.0838
Adi H, Young PM, Chan H-K, Salama R, Traini D (2010) Controlled release antibiotics for dry powder lung delivery. Drug Dev Ind Pharm 36(1):119–126. doi:10.3109/03639040903099769
Westerman EM, De Boer AH, Le Brun PPH, Touw DJ, Roldaan AC, Frijlink HW, Heijerman HGM (2007) Dry powder inhalation of colistin in cystic fibrosis patients: a single dose pilot study. J Cyst Fibros 6(4):284–292. doi:10.1016/j.jcf.2006.10.010
Hodson ME, Gallagher CG, Govan JRW (2002) A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur Respir J 20(3):658–664. doi:10.1183/09031936.02.00248102
Schuster A, Haliburn C, Döring G, Goldman MH; Freedom Study Group (2013) Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: a randomised study. Thorax 68(4):344–350. doi:10.1136/thoraxjnl-2012-202059
Doan TV, Couet W, Olivier JC (2011) Formulation and in vitro characterization of inhalable rifampicin-loaded PLGA microspheres for sustained lung delivery. Int J Pharm 414(1–2):112–117. doi:10.1016/j.ijpharm.2011.05.007
Cook RO, Pannu RK, Kellaway IW (2005) Novel sustained release microspheres for pulmonary drug delivery. J Control Release 104(1):79–90. doi:10.1016/j.jconrel.2005.01.003
Rukholm G, Mugabe C, Azghani AO, Omri A (2006) Antibacterial activity of liposomal gentamicin against Pseudomonas aeruginosa: a time–kill study. Int J Antimicrob Agents 27(3):247–252. doi:10.1016/j.ijantimicag.2005.10.021
Mugabe C, Azghani AO, Omri A (2005) Liposome-mediated gentamicin delivery: development and activity against resistant strains of Pseudomonas aeruginosa isolated from cystic fibrosis patients. J Antimicrob Chemother 55(2):269–271. doi:10.1093/jac/dkh518
Ré M-I (2006) Formulating drug delivery systems by spray drying. Dry Technol 24(4):433–446. doi:10.1080/07373930600611877
Varde NK, Pack DW (2004) Microspheres for controlled release drug delivery. Expert Opin Biol Ther 4(1):35–51. doi:10.1517/14712598.4.1.35
Liu W, Yang X-L, Ho WS (2011) Preparation of uniform-sized multiple emulsions and micro/nano particulates for drug delivery by membrane emulsification. J Pharm Sci 100(1):75–93. doi:10.1002/jps.22272
Manca ML, Mourtas S, Dracopoulos V, Fadda AM, Antimisiaris SG (2008) PLGA, chitosan or chitosan-coated PLGA microparticles for alveolar delivery? A comparative study of particle stability during nebulization. Colloids Surf B Biointerfaces 62(2):220–231. doi:10.1016/j.colsurfb.2007.10.005
Ravindra S, Varaprasad K, Narayana Reddy N, Vimala K, Mohana Raju K (2011) Biodegradable microspheres for controlled release of an antibiotic ciprofloxacin. J Polym Environ 19(2):413–418. doi:10.1007/s10924-011-0290-8
Mura S, Hillaireau H, Nicolas J, Le Droumaguet B, Gueutin C, Zanna S, Tsapis N, Fattal E (2011) Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int J Nanomedicine 6(1):2591–2605. doi:10.2147/IJN.S24552
Yang Y, Bajaj N, Xu P, Ohn K, Tsifansky MD, Yeo Y (2009) Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials 30(10):1947–1953. doi:10.1016/j.biomaterials.2008.12.044
O’Hara P, Hickey AJ (2000) Respirable PLGA microspheres containing rifampicin for the treatment of tuberculosis: manufacture and characterization. Pharm Res 17(8):955–961. doi:10.1023/a:1007527204887
Giovagnoli S, Blasi P, Schoubben A, Rossi C, Ricci M (2007) Preparation of large porous biodegradable microspheres by using a simple double-emulsion method for capreomycin sulfate pulmonary delivery. Int J Pharm 333(1–2):103–111. doi:10.1016/j.ijpharm.2006.10.005
Gupta V, Rawat A, Ahsan F (2010) Feasibility study of aerosolized prostaglandin E(1) microspheres as a noninvasive therapy for pulmonary arterial hypertension. J Pharm Sci 99(4):1774–1789. doi:10.1002/jps.21946
Ventura CA, Tommasini S, Crupi E, Giannone I, Cardile V, Musumeci T, Puglisi G (2008) Chitosan microspheres for intrapulmonary administration of moxifloxacin: Interaction with biomembrane models and in vitro permeation studies. Eur J Pharm Biopharm 68(2):235–244. doi:10.1016/j.ejpb.2007.05.011
Chowdary KPR, Rao YS (2004) Mucoadhesive microspheres for controlled drug delivery. Biol Pharm Bull 27(11):1717–1724. doi:10.1248/bpb.27.1717
Chen HY, Yuan M, Livermore DM (1995) Mechanisms of resistance to β-lactam antibiotics amongst Pseudomonas aeruginosa isolates collected in the UK in 1993. J Med Microbiol 43(4):300–309. doi:10.1099/00222615-43-4-300
Sharma R, Saxena D, Dwivedi AK, Misra A (2001) Inhalable microparticles containing drug combinations to target alveolar macrophages for treatment of pulmonary tuberculosis. Pharm Res 18(10):1405–1410. doi:10.1023/a:1012296604685
Ventura CA, Cannavà C, Stancanelli R, Paolino D, Cosco D, La Mantia A, Pignatello R, Tommasini S (2011) Gemcitabine-loaded chitosan microspheres. Characterization and biological in vitro evaluation. Biomed Microdevices 13(5):799–807. doi:10.1007/s10544-011-9550-6
Tuli RA, Dargaville TR, George GA, Islam N (2012) Polycaprolactone microspheres as carriers for dry powder inhalers: effect of surface coating on aerosolization of salbutamol sulfate. J Pharm Sci 101(2):733–745. doi:10.1002/jps.22777
Corrigan DO, Healy AM, Corrigan OI (2006) Preparation and release of salbutamol from chitosan and chitosan co-spray dried compacts and multiparticulates. Eur J Pharm Biopharm 62(3):295–305. doi:10.1016/j.ejpb.2005.09.008
Learoyd TP, Burrows JL, French E, Seville PC (2008) Modified release of beclometasone dipropionate from chitosan-based spray-dried respirable powders. Powder Technol 187(3):231–238. doi:10.1016/j.powtec.2008.02.015
Sakagami M, Kinoshita W, Sakon K, Sato J, Makino Y (2002) Mucoadhesive beclomethasone microspheres for powder inhalation: their pharmacokinetics and pharmacodynamics evaluation. J Control Release 80(1–3):207–218. doi:10.1016/s0168-3659(02)00034-2
Harsha S, Chandramouli R, Rani S (2009) Ofloxacin targeting to lungs by way of microspheres. Int J Pharm 380(1–2):127–132. doi:10.1016/j.ijpharm.2009.07.020
Jeong Y-I, Kim D-G, Seo D-H, Jang M-K, Nah J-W (2008) Multiparticulation of ciprofloxacin HCl-encapsulated chitosan microspheres using poly(dl-lactide-co-glycolide). J Ind Eng Chem 14(6):747–751. doi:10.1016/j.jiec.2008.05.004
Dillen K, Vandervoort J, Van den Mooter G, Verheyden L, Ludwig A (2004) Factorial design, physicochemical characterisation and activity of ciprofloxacin-PLGA nanoparticles. Int J Pharm 275(1–2):171–187. doi:10.1016/j.ijpharm.2004.01.033
Poyner EA, Alpar HO, Almeida AJ, Gamble MD, Brown MRW (1995) A comparative study on the pulmonary delivery of tobramycin encapsulated into liposomes and PLA microspheres following intravenous and endotracheal delivery. J Control Release 35(1):41–48. doi:10.1016/0168-3659(95)00017-3
Cystic Fibrosis Foundation (2012) Patient registry. Annual data report 2011. Available online at: http://www.cff.org/UploadedFiles/research/ClinicalResearch/2011-Patient-Registry.pdf. Accessed 11 March 2013
Cystic Fibrosis Foundation (2012) Drug development pipeline. Available online at: http://www.cff.org/research/DrugDevelopmentPipeline/. Accessed 8 January 2013
Acknowledgements
This work was supported by Fundação para a Ciência e a Tecnologia (FCT), Portugal, via the grant SFRH/BD/80307/2011.
Conflict of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gaspar, M.C., Couet, W., Olivier, JC. et al. Pseudomonas aeruginosa infection in cystic fibrosis lung disease and new perspectives of treatment: a review. Eur J Clin Microbiol Infect Dis 32, 1231–1252 (2013). https://doi.org/10.1007/s10096-013-1876-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-013-1876-y