
Abstract
Serotonin 2A receptor (HTR2A) gene was implicated to be associated with major depressive disorder (MDD) susceptibility due to its role of key neurotransmitter in many physiologic processes. A great number of related studies reported in different populations have emerged. The results of these studies, however, have been inconsistent and thereby definite conclusions are difficult to establish. With the cumulative data in recent years, it was necessary to carry out a comprehensive analysis of previous findings. Electronic databases were systematically searched for studies published before May 2013. Pooled odds ratios (OR) and 95 % confidence interval (CI) were estimated under three different genetic models. Subgroup and sensitivity analyses were also performed. A total of 21 studies, 3,299 patients and 4,092 controls, met the selection criteria. 15 studies included HTR2A T102C polymorphism (with a total of 2,409 patients and 3,130 controls), and 9 studies included HTR2A A-1438G polymorphism (with a total of 1,510 patients and 2,281 controls). Our results showed that no significant association of MDD susceptibility with T102C polymorphism was found in allelic analysis and genotypic analysis (For T vs. C: OR = 1.06, 95 % CI = 0.95–1.18, P = 0.307; For TT + TC vs. CC: OR = 1.07, 95 % CI = 0.90–1.28, P = 0.451; For TT vs. TC + CC: OR = 1.08, 95 % CI = 0.95–1.22, P = 0.235). With respect to A-1438G polymorphism, however, carriers with A allele tend to suffer from MDD (AA + AG vs. GG: OR = 1.20, 95 % CI = 1.02–1.43, P = 0.030). When stratified by race for T102C polymorphism and A-1438G polymorphism of the HTR2A, we found no significant association. In conclusions, our study suggests that the A allele of A-1438G polymorphism might play a role in susceptibility to MDD. On the contrary, T102C polymorphism does not seem to be capable of modifying MDD risk.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a pressing public health issues due to its high lifetime prevalence of approximately 15 % and its association with significant disability [1]. MDD is the second leading case of disease burden worldwide, and will be the first leading cause in high-income countries for disability-adjusted life years (DALY) in 2030 [2].
Epidemiological studies indicated that MDD is a disease involving genetic and environmental factors [3]. The total contribution of genetic factors in the origin of MDD is roughly 40 % [4]. Increasing evidences from clinical and pharmacological studies indicate that altered serotonergic neural transmission is a susceptible factor for MDD [5], which has been discussed in detail elsewhere [6, 7]. Several postmortem studies have illustrated that an increase in the number of the serotonin 2A receptor (HTR2A) is associated with the frontal cortex of depressed patients and suicide victims [8, 9].
There are many polymorphisms for HTR2A gene, located on the long arm of chromosome 13 [10], of which the most extensively investigated single nucleotide polymorphisms (SNPs) are A-1438G and T102C corresponding to NCBI dbSNP cluster IDs, rs6311 and rs6313, respectively. One study shows an association between C allele of the T102C polymorphism and MDD [11] and another study find that T allele of the T102C polymorphism is associated with an increased risk for MDD [12]. Several studies show negative associations between the T102C polymorphism and MDD [13–15]. Furthermore, the A-1438G polymorphism of the HTR2A gene is reported to be significantly associated with MDD [16], while there are also negative associations between this SNP and MDD [13, 17].
A meta-analysis published by Anguelova in 2003 [18], has indicated that the T102C polymorphism is not associated with MDD. However, the study ignored articles in Chinese databases (CNKI, CBM, and Wan Fang) and only included seven studies (five in Europe and two in Asia). Furthermore, the subgroup and sensitivity analyses were not performed in their study. To illustrate the effects of the two SNPs on the MDD, we conducted a meta-analysis to examine the association of the variation of HTR2A gene with MDD.
Methods
Literature identification
A comprehensive literature search was carried out in PubMed, Chinese biomedical database (CBM), Chinese national knowledge infrastructure (CNKI), and Wan fang (Chinese) database to collect the articles of case–control studies or cohort studies on associations between HTR2A gene and MDD susceptibility before May 2013. We also performed searching based on www.baidu.com and www.google.com to identify additional studies. The PubMed search was run using the mesh terms: [‘Receptor, Serotonin, 5-HT2A’ [Mesh], ‘5-Hydroxytryptamine 2A’, or ‘5-HT 2A’, or ‘HTR2A’] and [‘depression’, ‘depressive disorder’, or ‘depressive disorders’]. All references cited in these studies and published reviews were examined to identify additional works. The search results were limited to English language and Chinese language publications.
Inclusion criteria
Eligible studies had to meet all of the following criteria: (1) they had to be case–control or cohort studies that investigated the association between the T102C or A-1438G genotype and MDD; (2) they should have presented sufficient data to calculate the odds ratio (OR) with 95 % confidence interval (95 % CI); (3) they should have diagnosed MDD patients according to the ICD, DSM-IV criteria or Chinese classification of mental disorders (CCMD) systems; (4) they should have used healthy individuals as controls; (5) the distribution of the genotypes in control groups was in the Hardy–Weinberg equilibrium; (6) the subjects were humans; and (7) they were independent from one another. Analysis based on the same sets of data was excluded. In such cases, only the larger sample was accounted for. Meeting abstracts, case reports, editorials, and review articles were excluded.
Data extraction and synthesis
Data were extracted by two of the authors independently using a standardized data extraction form. Discrepancies were resolved by discussion and the decision was made by the third authors if consensus was not achieved. The title and abstract of all potentially relevant articles were screened to determine their relevance. Full articles were also scrutinized if the title and abstract were ambiguous. The following information was collected from each study: name of the first author, year of publication, study design, sample size, geographical location, ethnicity of participants, definition and numbers of cases and controls, genotype information, and outcome.
Statistical analyses
Data from each study were used to construct a two-by-two table in which subjects were classified by diagnostic category and type of allele. Statistical heterogeneity among studies was assessed with the Q and I2 statistics. On condition that heterogeneity was found, the random effects model was adopted to combine each samples, and to calculate the OR and the corresponding 95 % confidence interval (CI); otherwise, the fixed effects model was adopted. The significance of the pooled OR was determined by the z test. Publication bias was investigated with the funnel plot, in which the standard error of log OR of each study was plotted against its OR. Funnel-plot asymmetry was further assessed by the method of Egger’ liner regression test [19]. For the sensitivity analysis, a single study involved in the meta-analysis was deleted each time to reflect the influence of the individual data set to the pooled OR. Analyses were performed using the software Stata version 11.2 (Stata Corp LP, College Station, TX, USA). All the P values were two-sided. A P value less than 0.05 was considered statistically significant.
Results
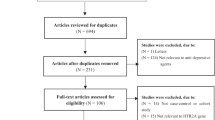
Study selection
The combined search yielded 554 references for studies with MDD in PubMed, 28 references in CBM, and correspondingly 21 references in CNKI. Another two studies [20, 21] were identified by tracing back literatures. After searching the medical subject heading terms and abstracts, 26 articles [11–17, 20–38] were obtained. Articles were then confirmed on the inclusion criteria. Five studies did not meet the inclusion criteria, of which one study [22] was excluded because it was about symptomatology; one study [23] was excluded for the case group from patients with depression after stroke; two studies [24, 25] for using different diagnosis standards, the Center for Epidemiologic Studies Depression Scale (CES-D) and the Mood and Feelings Questionnaire (SMFQ), respectively; one [26] for studying the association between the -759,-697 allele and -759/-697 genotype of the HTR2A gene and depression. Finally, 21 articles [11–17, 20, 21, 27–38] met the inclusion criteria, including 3,299 patients and 4,092 controls. Of which eight [30–35, 37, 38] were written in Chinese. The studies are described in Table 1.
Meta-analysis
The T102C polymorphism of HTR2A gene
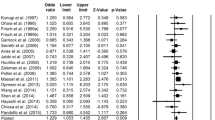
Among all 15 articles [11–15, 27–36] that investigated HTR2A T102C polymorphism and MDD, a total of 2,409 patients and 3,130 controls were included. The level of heterogeneity between studies varied, so both fixed-effect models and random-effect models were used. Finally, we failed to observe a significant association between HTR2A T102C polymorphism and MMD with any genetic model (For T vs. C: P = 0.307; For TT + TC vs. CC: P = 0.451; For TT vs. TC + CC: P = 0.235). Furthermore, we divided the studies by ethnicity; results showed no statistically significant differences in Caucasians and Asians (P > 0.05) (Table 2). The sensitivity analysis did not change the pattern of results. But in Caucasian, publication bias was observed in the genotypic analysis when the “C” genotypics were combined (t = −4.37, P = 0.007). The forest and funnel plots are presented in Figs. 1 and 2.

Forest plot for the overall association between HTR2A T102C polymorphism and major depressive disorder

Egger’s linear regression test for publication bias analysis of association studies between HTR2A T1O2C polymorphism and major depressive disorder
However, the frequency of the T allele of HTR2A T102C polymorphism was slightly higher in Asian patients and controls (54.41 and 52.04 %, respectively) than in Caucasian patients and controls (44.78 and 42.87 %, respectively).
The A-1438G polymorphism of HTR2A gene
Among all 9 articles on HTR2A A-1438G polymorphism and MDD [13, 16, 17, 20, 21, 33, 36–38], a total of 1,510 patients and 2,281 controls were included. Significant heterogeneity was identified for the allelic and genotypic analysis of A-1438G (for A vs. G: P = 0.003; for AA vs. AG + GG: P = 0.003) (Table 2). For the A-1438G allelic analysis and the genotypic analysis with G allele combined, the results were non-significant (P = 0.110, P = 0.318, respectively). However, the genotypic analysis with A allele combined showed significant association (P = 0.030) (Table 2). In the further analyses of stratifying studies by ethnicity, no significant association was found in Asians and Caucasians (P > 0.05) (Table 2). There was no publication bias according to Egger’s test, with P values >0.05 (for A vs. G: t = 0.88, P = 0.409; for AA + AG vs. GG: t = 1.50, P = 0.178; for AA vs. AG + GG: t = 0.22, P = 0.835). The forest plot was presented in Fig. 3.

Forest plot for the overall association between HTR2A A–1438G polymorphism and major depressive disorder
However, sensitivity analyses indicated that after excluding the study of Choi et al. [16], no study heterogeneity was observed (for A vs. G: P = 0.154; for AA + AG vs. GG: P = 0.486; for AA vs. AG + GG: P = 0.230). In addition, the patterns of statistical results from the fixed effect model were altered (for A vs. G: P = 0.001; for AA + AG vs. GG: P = 0.005; for AA vs. AG + GG: P = 0.009).
Discussion
Similar to the previous meta-analysis conducted by Anguelova et al. [18] in 2003, which reported that the HTR2A T102C polymorphism was not directly associated with depressive disorders, we found no significant association. Considering the influence of ethnicity, we also performed a subgroup analysis stratified by ethnicity, Asian and Caucasian. The results of subgroup analyses also reported non-significant. Compared to the meta-analysis published by Anguelova, our study included another nine new studies which involved three studies published after 2003 [13, 14, 36] and six papers from Chinese databases [30–35]. Therefore, our study has higher statistical power than the previous meta-analysis conducted by Anguelova et al. [18].
Furthermore, we examined the relationship between the A-1438G polymorphism of the HTR2A gene and MDD. The results indicated that carriers with A allele of A-1438G were associated with MDD (AA + AG vs. GG: OR = 1.20, 95 % CI = 1.02–1.43, P = 0.030), and after excluding the study of Choi et al. [16], we obtained significant associations between the A-1438G polymorphism and MDD with all models (P < 0.01). It was different from a previous meta-analysis conducted by Jin et al. [39]. who reported that no significant association between the HTR2A A-1438G polymorphism and the risk of MDD was observed. Compared with the data from Jin, our study included one more study of Li et al. [33] and excluded three studies, of which two studies of Enoch et al. [40] and Molnar et al. [41] were excluded because they examined the association of the A-1438G polymorphism with seasonal affective disorder (SAD); the study of Frisch et al. [29] was excluded because it examined the association of the T102C polymorphism with MDD. Thus our results about the association between A-1438G polymorphism and MDD may be more statistically convincing. Although our results indicated that carriers with A allele tend to suffer from MDD, the P value obtained (P = 0.030 for AA + AG vs. GG) was very close to the level of marginal significance. Therefore, further studies with large sample size or high statistical power are required to reach a definite conclusion.
With the rapid development of technological advances in genomics, it is possible to genotype 500,000–1 million SNPs across the genome in cases and healthy controls now. This genome-wide association study (GWAS) design has the advantage that no genes are preselected (as is the case in candidate gene studies), and robust findings might identify new pathways involved in MDD. Thus far, eight GWAS for MDD have been published [42–49]. To identify robust and replicable associations, the psychiatric genomics consortium (PGC) conducted a meta-analysis of genome-wide genetic data for MDD, which contained more than 1.2 million autosomal and X chromosome SNPs, and the SNPs rs6313(T102C) and rs6311(A-1438G) in HTR 2A gene were included in [50]. The results showed that no SNPs achieved genome-wide significance in the MDD discovery phase, the MDD replication phase or in preplanned secondary analyses (by sex, recurrent MDD, recurrent early-onset MDD, age of onset, pre-pubertal onset MDD or typical-like MDD from a latent class analyses of the MDD criteria). However, there has been considerable speculation that gene-environment interactions are particularly salient for MDD. Larger study cohorts characterized for genetic and environmental risk factors accumulated prospectively are likely to be needed to dissect more fully the etiology of MDD.
The characteristic of meta-analysis is to combine comparable studies to increase the sample size and statistical power and draw a more reliable result. However, the result of meta-analysis may be influenced by some factors such as publication bias, method of sampling, different genetic backgrounds of subjects, different protocols and quality of analysis. We obeyed the inclusion criteria strictly to reduce selection bias. Funnel plot and Egger’s linear regression test were used to assess publication bias. In addition, our inclusion of non-English language reports was important in minimizing a major potential threat to the validity of any meta-analysis, the related threat of a language bias, and the impact of different genetic background was lessened by means of subgroup analysis. Finally, the sensitivity analysis had been performed to confirm the reliability and stability of this meta-analysis.
There are several limitations to this meta-analysis. Firstly, Publication bias is a major problem in performing meta-analysis and it might occur even though it was not found in statistical tests. Secondly, observational studies are susceptible to various biases (e.g., recall bias in case–control studies) because of their retrospective nature. Thirdly, the quality of the included studies was heterogeneous. Finally, the choice of control participants in case–control studies may distort the results because hospital-based controls may not be as representative as population-based controls, although no evidence of the effect of control population was detected in this meta-analysis.
To conclude, this meta-analysis suggests that the A allele of A-1438G polymorphism might play a role in susceptibility to MDD. On the contrary, T102C polymorphism does not seem to be capable of modifying MDD risk. However, we must consider that MDD is a polymorphic and multifactorial disorder. Larger studies in selected populations with different environmental backgrounds and/or other risk gene polymorphism are needed to provide more reliable evidence for the relationship between HTR2A gene and MDD.
References
Moussavi S, Chatterji S, Verdes E, Tandon A, Patel V, Ustun B (2007) Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet 370:851–858
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442
Duffy A, Grof P, Robertson C, Alda M (2000) The implications of genetics studies of major mood disorders for clinical practice. J Clin Psychiatry 61:630–637
Sullivan PF, Neale MC, Kendler KS (2000) Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 157:1552–1562
Owens MJ, Nemeroff CB (1998) The serotonin transporter and depression. Depress Anxiety 8(suppl 1):5–12
Levinson DF (2006) The genetics of depression: a review. Biol Psychiatry 60:84–92
Murphy DL, Lerner A, Rudnick G, Lesch KP (2004) Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Mol Interv 4:109–123
Arora RC, Meltzer HY (1989) Serotonergic measures in the brains of suicide victims: 5-HT (2A) binding sites in the frontal cortex of suicide victims and control subjects. Am J Psychiatry 146:730–736
Yates M, Leake A, Candy JM, Fairbairm AF, McKeith IG, Ferrier IN (1990) 5HT 2A receptor changes in major depression. Biol Psychiatry 27:489–496
Williams J, Spurlock G, McGuffin P, Mallet J, Nothen MM, Gill M, Aschauer H, Nylander PO, Macciardi F, Owen MJ (1996) Association between schizophrenia and T102C polymorphism of the 5-hydroxytryptamine type 2a-receptor gene, European Multicentre Association Study of Schizophrenia (EMASS) Group. Lancet 347:1294–1296
Du L, Bakish D, Lapierre YD, Ravindran AV, Hrdina PD (2000) association of polymorphism of serotonin 2A receptor gene with suicidal ideation in major depressive disorder. Am J Med Genet (Neuropsychiatric Genetics) 96:56–60
Zhang HY, Ishigaki T, Tani K, Chen K, Shih JC, Miyasato K, Ohara K, Ohara K (1997) Serotonin 2A receptor gene polymorphism in mood disorders. Biol Psychiatry 41:768–773
Kishi T, Kitajima T, Tsunoka T, Ikeda M, Yamanouchi Y, Kinoshita Y, Kawashima K, Okochi T, Okumura T, Inada T, Ozaki N, Iwata N (2009) Genetic association analysis of serotonin 2A receptor gene (HTR2A) with bipolar disorder and major depressive disorder in the Japanese population. Neurosci Res 64:231–234
Khait VD, Huang YY, Zalsman G, Oquendo MA, Brent DA, Harkavy-Friedman JM, Mann JJ (2005) Association of serotonin 5-HT2A receptor binding and the T102C polymorphism in depressed and healthy Caucasian subjects. Neuropsychopharmacology 30:166–172
Minov C, Baghai TC, Schüle C, Zwanzger P, Schwarz MJ, Zill P, Rupprecht R, Bondy B (2001) Serotonin-2A-receptor and –transporter polymorphisms: lack of association in patients with major depression. Neurosci Lett 303:119–122
Choi MJ, Lee HJ, Lee HJ, Ham BJ, Cha JH, Ryu SH, Lee MS (2004) Association between major depressive disorder and the –1438A/G polymorphism of the serotonin 2A receptor gene. Neuropsychobiology 49:38–41
Tencomnao T, Thongrakard V, Phuchana W, Sritharathikhun T, Suttirat S (2010) No relationship found between -1438A/G polymorphism of the serotonin 2A receptor gene (rs6311) and major depression susceptibility in a northeastern Thai population. Genet Mol Res 9:1171–1176
Anguelova M, Benkelfat C, Turecki G (2003) A systematic review of association studies investigating genes coding for serotonin receptors and the serotonin transporter: i affective disorders. Mol Psychiatry 8:574–591
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315:629–634
Ohara K, Nagai M, Tsukamoto T, Tani K, Suzuki Y, Ohara K (1998) 5-HT2A receptor gene promoter polymorphism—1438G/A and mood disorders. Neuroreport 9:1139–1141
Bonnier B, Gorwood P, Hamon M, Sarfati Y, Boni C, Hardy- Bayle MC (2002) Association of 5-HT2A receptor gene polymorphism with major affective disorders: the case of a subgroup of bipolar disorder with low suicide risk. Biol Psychiatry 51:762–765
Christiansen L, Tan Q, Iachina M, Bathum L, Kruse TA, McGue M, Christensen K (2007) Candidate gene polymorphisms in the serotonergic pathway: influence on depression symptomatology in an elderly population. Biol Psychiatry 61:223–230
Kim JM, Stewart R, Bae KY, Kim SW, Kang HJ, Shin IS, Kim JT, Park MS, Kim MK, Park SW, Kim YH, Kim JK, Cho KH, Yoon JS (2012) Serotonergic and BDNF genes and risk of depression after stroke. J Affect Disord 136:1–8
Jansson M, Gatz M, Berg S, Johansson B, Malmberg B, McClearn GE, Schalling M, Pedersen NL (2003) Association between depressed mood in the elderly and a 5-HTR2A gene variant. Am J Med Genet B (Neuropsychiatr Genet) 120B:79–84
Eley TC, Sugden K, Corsico A, Gregory AM, Sham P, McGuffin P, Plomin R, Craig IW (2004) Gene-environment interaction analysis of serotonin system markers with adolescent depression. Mol Psychiatry 9:908–915
Lu Z, Cai J, Jiang SD, Wang DX, Li X, Yao PF (2005) Analysis of association between polymorphism of serotonin 2A, 2C receptor gene and treatment-resistant depression. J Clin Psychol Med (Chinese) 15:193–195
Arias B, Gutierrez B, Pintor L, Gasto C, Fananas L (2001) Variability in the 5-HT2A receptor gene is associated with seasonal pattern in major depression. Mol Psychiatry 6:239–242
Bondy B, Kuznik J, Baghai T, Schüle C, Zwanzger P, Minov C, de Jonge S, Rupprecht R, Meyer H, Engel RR, Eisenmenger W, Ackenheil M (2000) Lack of association of serotonin-2A receptor gene polymorphism (T102C) with suicidal ideation and suicide. Am J Med Genet (Neuropsychiatr Genet) 96:831–835
Frisch A, Postilnick D, Rockah R, Michaelovsky E, Postilnick S, Birman E, Laor N, Rauchverger B, Kreinin A, Poyurovsky M, Schneidman M, Modai I, Weizman R (1999) Association of unipolar major depressive disorder with genes of the serotonergic and dopaminergic pathways. Mol Psychiatry 4:389–392
Liu JL, Wang YF, Sun N, Du QR, Lang XE, Li X (2008) An association study on the T102C polymorphism of the serotonin 2A receptor gene and major depression in northern china. Chin J Nerv Ment Dis (Chinese) 34:181–182
Wang GJ, Zhong AF, Zhang LY, Wang HL, Zhao HQ, Yu HY (2002) A study on the association between gene polymorphism of serotonin 2A receptor and depression. Sichuan Jing Shen Wei Sheng Za Zhi (Chinese) 15:147–148
Cai J, Lu Z, Jiang SD, Li X, Wang DX, Yao PF (2004) Association of 5-HT2A receptor gene polymorphisms and treatment in depression. J Clin Psychol Med (Chinese) 14:129–130
Li XL, Fan CH, Jia FJ, Li LH, Tan HH, Shan ZX (2009) An association study on polymorphisms of 5-HT2A gene and major depression. J Neurosci Ment Health (Chinese) 9:11–14
Xu GQ, Li YF (2006) The relationship between 5-HT2A polymorphism and depression an d suicide behavior. Shanghai Archives of Psychiatry (Chinese) 18:161–163
Jiang KD, Jiang SD, Wang HX, Yang XM, Wang HF, Lin CC (1999) An interaction between 5-HT 2A receptor gene and MAOA gene in unipolar depression patients. Chin J Nerv Ment Dis (Chinese) 25:90–92
Illi A, Setala-Soikkeli E, Viikki M, Poutanen O, Huhtala H, Mononen N, Lehtimäki T, Leinonen E, Kampman O (2009) 5-HTR1A, 5-HTR2A, 5-HTR6, TPH1 and TPH2 polymorphisms and major depression. Neuro Report 20:1125–1128
Yu Y, Zhao JP, Wu RR, Xun GL, Fang MS (2009) Association study between major depression and the-1438A/G polymorphism of serotonin receptor 2A gene. J Int Psychiatry 36:65–68
Zhu YZ, Zhang Y, Ma H, Xie YF, Jiang WY, Sun GW (2010) 5-HTR2A gene-1438A/G polymorphism and effect of fluoxetine in major depressive disorder patients. Chin Gen Pract 13:1431–1434
Jin CH, Xu WW, Yuan JM, Wang GQ, Cheng ZH (2013) Meta-analysis of association between the -1438A/G (rs6311) polymorphism of the serotonin 2A receptor gene and major depressive disorder. Neurol Res 35:7–14
Enoch MA, Goldman D, Barnett R, Sher L, Mazzanti CM, Rosenthal NE (1999) Association between seasonal affective disorder and the 5-HT2A promoter polymorphism, -1438G/A. Mol Psychiatry 4:89–92
Molnar E, Lazary J, Benko A, Gonda X, Pap D, Mekli K, Juhasz G, Kovacs G, Kurimay T, Rihmer Z, Bagdy G (2010) Seasonality and winter-type seasonal depression are associated with the rs731779 polymorphism of the serotonin-2A receptor gene. Eur Neuropsychopharmacol 20:655–662
Rietschel M, Mattheisen M, Frank J, Treutlein J, Degenhardt F, Breuer R et al (2010) Genome-wide association, replication, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry 68:578–585
Sullivan P, de Geus E, Willemsen G, James MR, Smit JH, Zandbelt T et al (2009) Genomewide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry 14:359–375
Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA et al (2011) Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry 16:193–201
Muglia P, Tozzi F, Galwey NW, Francks C, Upmanyu R, Kong XQ et al (2010) Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry 15:589–601
Wray N, Pergadia M, Blackwood D, Penninx B, Gordon S, Nyholt D et al (2011) Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 17:36–48
Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K et al (2011) The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron 70:252–265
Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K et al (2010) Genome-wide association study of recurrent major depression in the UK population. Am J Psychiatry 167:949–957
Shyn SI, Shi J, Kraft JB, Potash JB, Knowles JA, Weissman MM et al (2011) Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol Psychiatry 16:202–215
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium (2012) A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. doi:10.1038/mp.2012.21
Acknowledgments
The present study was supported by National Natural Science Foundation of China (81172763).
Conflicts of interest
There are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
X. Zhao and L. Sun contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhao, X., Sun, L., Sun, YH. et al. Association of HTR2A T102C and A-1438G polymorphisms with susceptibility to major depressive disorder: a meta-analysis. Neurol Sci 35, 1857–1866 (2014). https://doi.org/10.1007/s10072-014-1970-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-014-1970-7