
Abstract
Chronic cerebral hypoperfusion contributes to a cognitive decline related to brain disorders. Its experimental model in rats is a permanent bilateral common carotid artery occlusion (2VO). Overstimulation of the glutamatergic system excitotoxicity due to brain energetic disturbance in 2VO animals seems to play a pivotal role as a mechanism of cerebral damage. The nucleoside guanosine (GUO) exerts extracellular effects including antagonism of glutamatergic activity. Accordingly, our group demonstrated several neuroprotective effects of GUO against glutamatergic excitotoxicity. Therefore, in this study, we evaluated a chronic GUO treatment effects in rats submitted to 2VO. We evaluated the animals performance in the Morris water maze and hippocampal damage by neurons and astrocytes immunohistochemistry. In addition, we investigated the cerebrospinal fluid (CSF) brain derived neurotrophic factor (BDNF) and serum S100B levels. Additionally, the purine CSF and plasma levels were determined. GUO treatment did not prevent the cognitive impairment promoted by 2VO. However, none of the 2VO animals treated with GUO showed differences in the hippocampal regions compared to control, while 20% of 2VO rats not treated with GUO presented loss of pyramidal neurons and increased glial labeling cells in CA1 hippocampal region. In addition, we did not observe differences in CSF BDNF nor serum S100B levels among the groups. Of note, both the 2VO surgery and GUO treatment changed the purine CSF and plasma profile. In conclusion, GUO treatment did not prevent the cognitive impairment observed in 2VO animals, but our data suggest that GUO could be neuroprotective against hippocampal damage induced by 2VO.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vascular dementia (VD), which is a group of diseases with heterogeneous pathological conditions and physiopathological mechanisms, is the second most common cause of dementia associated with Alzheimer’s disease and accounts for 10–50% of all dementias [1, 2]. Chronic cerebral hypoperfusion is considered a factor that contributes to memory dysfunction in neurological diseases such as VD [3, 4]. Moreover, the degree of cerebral hypoperfusion has been suggested as a predictive biomarker of the gradual transition from a mild cognitive impairment to the Alzheimer’s disease [5].
Permanent bilateral occlusion of the common carotid arteries (also denominated two vessels occlusion, 2VO) in rats is used as a chronic cerebral hypoperfusion model [6]. This procedure results in significant reduction of cerebral blood flow [6–9] and can cause progressive, long-lasting cognition deficits and neuronal damage resembling the effects observed in VD [9, 10]. 2VO in rats provides a useful model to understand the pathophysiology of chronic cerebral vascular disorders and to screen drugs with potential therapeutic value in VD.
The chain of events that eventually leads to neuronal cell death in chronic cerebral hypoperfusion begins with neuronal energy failure due to the blood flow reduction and the consequent oxygen and glucose deficiency [6, 11–13]. The disturbance of the energetic metabolism leads accumulation of extracellular glutamate [14, 15], the main excitatory neurotransmitter in the central nervous system, essential for the brain function [14, 16, 17]. However, overstimulation of the glutamatergic system (excitotoxicity), which increases the calcium influx triggers several intracellular processes, such as proteolytic hydrolysis, lipid peroxidation, and generation of reactive oxygen species, causing neuronal death [16, 18]. Thus, excitotoxicity has been proposed as a mechanism of neuronal damage impairing cellular energetics, as observed in hypoperfusion [19, 20].
The nucleoside guanosine (GUO) exerts various extracellular signaling effects, such as trophic effects on neural cells [21–26] and in vitro and in vivo antagonism of the glutamatergic system [27–29]. Accordingly, our group has demonstrated by several works that GUO is neuroprotective in different in vitro and in vivo experimental models of glutamatergic excitotoxicity [27, 28, 30–37], including ischemic insults [38, 39]. The GUO mechanism of action is not fully understood, however, our group demonstrated that GUO stimulates glutamate uptake by cultured astrocytes and brain slices [40–44], a physiological process that prevent glutamate toxicity.
Therefore, considering the involvement of excitotoxicity in the 2VO model and the neuroprotective potential of GUO, the aim of the present study was to investigate the effects of GUO in rats submitted to 2VO. For this purpose, we treated the animals during the first 6 weeks after the 2VO surgery (the period where the cerebral blood flow is highly reduced [12, 45]) with orally chronic GUO administration. After 6 weeks, some animals were sacrificed for purine cerebrospinal fluid (CSF) and plasma level analysis. Furthermore, we performed behavioral and/or histological analysis 6 months postoperative in another rats since significant time-dependent changes in the neural cell markers had been reported to occur until that period [6, 46]. Accordingly, we evaluated the spatial memory performance of the rats in the Morris water maze task and the hippocampal histology. Moreover, we also investigated the CSF brain derived neurotrophic factor (BDNF) levels and the serum S100B levels as possible markers of brain insult.
Materials and methods
Animals
Male adult Wistar rats (90–100 days old, weighing 300–350 g) were kept on a 12-h light/dark cycle (light on at 7:00 a.m.) at constant temperature of 22 ± 1°C. They were housed in plastic cages (5 per cage) with water and commercial food ad libitum. All behavioral tasks were conducted between 9:00 a.m. and 5:00 p.m. All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals adopted by the National Institute of Health (USA) and with the Federation of Brazilian Societies for Experimental Biology (FESBE), and were approved by the Research Ethics Committee of Universidade Federal do Rio Grande do Sul.
Surgery procedure
Chronic cerebral hypoperfusion was performed (40 animals) by a modified protocol of permanent bilateral occlusion of the common carotid arteries (2VO) [47]; additionally, 40 animals served as sham-operated controls (SHAM). Rats were anesthetized with halothane. The common carotid arteries were exposed via a neck ventral midline incision, separated from their sheaths and vagal nerves. Rats were submitted to the modified 2VO protocol: carotids were permanently occluded with 5-0 silk suture with a 1-week interval between interventions, the right common carotid being the first to be assessed and the left one being occluded 1 week later. Sham-operated controls received the same surgical procedures without carotid artery occlusion.
Treatment and groups
Rats received water (for control group) or GUO solution (0.5 mg/ml) for 6 weeks ad libitum in the bottle water immediately from the 2VO surgery. The GUO dose was chosen based on a previously study, which demonstrated that it was neuroprotective against seizures induced by quinolinic acid (Vinadé et al. 2003). The water consumption and body weight were monitored during the period of treatment every 2 days. The animals were randomly assigned to four different groups: sham-operated animals receiving water (SHAM-CT) or GUO (SHAM-GUO) and 2VO operated animals receiving water (2VO-CT) or GUO (2VO-GUO). The purine CSF and plasma level were evaluated after the treatment. The behavioral, histological and proteins analysis were performed 6 months postoperative.
CSF, serum and plasma sampling
CSF and blood (plasma or serum) were collected in two different times after the surgery for different parameters analysis.
Six weeks postoperative, 10 animals per group were sacrificed for purine CSF and plasma level measurements. The rats were anesthetized with sodium thiopental (40 mg/kg, 1 mL/kg, i.p.), and whole blood was obtained through a retrobulbar venous plexus puncture using a capillary tube. Plasma was separated by centrifugation at 3,000g for 10 min RT in a sodium-citrate tube. After, the animals were then positioned in a stereotaxic holder for CSF collection from the cisterna magna. The puncture was performed using an insulin syringe (27 gauge 9 1/200 length). The CSF were then centrifugated at 3,000g for 10 min 4°C to obtain a CSF cellfree supernatants. The CSF and plasma samples were frozen (−80°C) until analysis.
One day after the conclusion of the behavioral study (approximately 6 months postoperative), the CSF and serum of 40 animals (10 per group) were obtained for BDNF and/or S100B quantification, respectively. First, the rats were anesthetized and the CSF were collected accordingly reported above. After, rats were then removed from the stereotaxic apparatus and placed in a flat place; whole blood was obtained through an intracardiac puncture using a 0.37-mm diameter needle that was inserted into the intercostal space above the sternum. Serum was separated by centrifugation at 3,000g for 10 min RT. CSF and serum samples were frozen (−20°C) until analysis.
HPLC procedure
High-performance liquid chromatography (HPLC) was performed to measure the concentration of purines. The measurement was done according to previously determined guidelines [28]. It measured the CSF concentrations of the following purines: adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), adenosine (ADO), guanosine triphosphate (GTP), guanosine diphosphate (GDP), guanosine monophosphate (GMP), guanosine (GUO), inosine (INO), hypoxanthine (HIPOX), xanthine (XAN), and uric acid (UA). Analyses were performed with the Shimadzu Class-VP chromatography system, consisting of a quaternary gradient pump with vacuum degassing and piston desalting modules, Shimadzu SIL-10AF autoinjector valve with 50 mL loop and a UV detector (Shimadzu, Kyoto, Japan). Separations were achieved on a Supelco C18 250 ± 4.6 mm, 5-mm particle size column (Supelco, St Louis, MO, USA). The mobile phase flowed at a rate of 1.2 mL/min and the column temperature was 24°C. Buffer composition remained unchanged (A: 150 mmol/L phosphate buffer, pH 6.0, containing 150 mmol/L potassium chloride; B: 15% acetonitrile in buffer A). The gradient profile was modified to the following content of buffer B in the mobile phase: 0% at 0.00 min, 2% at 0.05 min, 7% at 2.45 min, 50% at 10.00 min, 100% at 11.00 min, and 0% at 12.40 min. Samples of 10 mL were injected into the injection valve loop. Absorbance was read at 254 nm. CSF and plasma concentrations of purines are expressed as mean ± SEM in micromoles.
Morris water maze
Cognitive function was evaluated in the Morris water maze task [48]. The maze consisted of a black circular pool (200 cm diameter, 60 cm height) filled to a depth of 40 cm with water (22 ± 1°C). A circular platform (10 cm diameter) was submerged 2 cm below the surface of the water and hidden from the rat’s view. Four points, equally spaced along the circumference of the pool, were arbitrarily assigned as North, South, East and West. The pool, therefore, was divided into four quadrants (Northeast, Southeast, Southwest, and Northwest). These points served as the starting positions where the rats were gently lowered into the water, with its head facing the wall of the water maze. We submitted the rats to a reference memory protocol for spatial memory analyzes.
Reference memory protocol
In this protocol, 10 rats per group received 5 training sessions (one session/day) and a probe trial in the 6th day. Each session consisted of four trials with a 15-min inter-trial interval. The location of the escape platform was fixed throughout training in the middle of the NE quadrant, 30 cm from the wall. A trial began when the rat was placed in the water at one of the 4 starting positions, randomly chosen, facing the wall. The order of the starting position varied in each trial and any given sequence was not repeated. A trial ended when the rat escaped onto the platform, and the escape latency for each trial was recorded. When the animal did not succeed, it was gently guided to the platform and left on it for 10 s and the escape latency was recorded as 60 s. Rats were dried and returned to their home cages after each trial. The mean escape latency of daily trials was then calculated.
Probe trial
In this 1-day test, each rat was subjected to a probe trial (60 s) without the platform. The latency to reach the original platform position, the number of crossings over that place and the time spent on the target quadrants were measured.
Sessions were recorded with a video acquisition system. Videotapes were used by a trained observer using dedicated software (ANY-maze®). Videos were subsequently placed in randomized order in a separate ANY-maze protocol to be scored by a trained observer blind to the experimental condition using a keyboard-based behavioral tracking system.
Quantification of CSF BDNF
BDNF levels in CSF were measured by anti-BDNF sandwich-ELISA, according to the datasheet from DuoSet kit (R&D Systems, Inc, USA) in a Spectra Max M5 molecular Devices (USA). Microtiter plates (96-well flat-bottom) were coated 2 h with samples (n = 10 per group) and reference curve standards (ranging from 23 to 1,500 pg/mL BNDF). The plates were then washed 4 times with PBS + Tween 0.05% and a diluted biotinylated mouse anti-human BDNF monoclonal (1:1,000) was added to each well and incubated for 2 h at room temperature. After this time, the plates were washed 4 times and then a diluted streptavidin-horseradish peroxidase (HRP) conjugate solution was added to each well and incubated at room temperature for 20 min. Wells were then washed four times before adding TMB chromogen (Tetramethylbenzidine) and maintained at room temperature for 20 min before the addition of stop solution. The amount of BDNF was determined at 450 nm and expressed as ng/mL.
Quantification of serum S100B
Serum S100B concentrations were measured using an enzyme linked immunosorbent assay (Diasorin® S100 ELISA Kit) in a Spectra Max M5 molecular Devices (USA). Calibrators and serum samples (100 μL—n = 10 per group) were incubated in a plate previously coated with anti-S100B antibody. The S100 ELISA was a two-site, one-step, enzyme linked immunosorbent assay. In the assay calibrators, controls and serum samples react simultaneously with two solid phase capture antibodies and a detector antibody conjugated with HRP during the incubation in the microtiter wells for 2 h. After a washing step (with PBS + Tween 0.05%) a TMB chromogen was added and the reaction was allowed to proceed for 15 min. The enzyme reaction was stopped by adding stop solution and the absorbance was measured at 450 nm. S100B concentrations were derived by comparison with the calibration curve based on the total absorbance for each given calibrator provided with the assay. All determinations were carried out within the same experiment. The S100B calibration curve is cubic spline up to 5 μg/L, and the CVs for duplicates across the entire concentration range for the calibrators and samples were 5%. The detection limit of the assay is 0.03 μg/L. The results are expressed as ng/mL.
Histological analysis
After CSF and serum sampling, the animals were injected with 400 IU of heparin (n = 10 per group). They were then submitted to transcardiac perfusion with 0.9% saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer pH 7.4. The brains were removed and post-fixed in the same solution at room temperature for 4 h. For the morphological analysis, brains were cryoprotected with a 30% sucrose solution for 2 days. Coronal sections (50 μm) were obtained using a Vibratome (Leica, Germany).
One section in six random series was collected for immunohistochemistry. Immunohistochemical detection was carried out in the sections with all fields of the hippocampus (an area of the hippocampus extending between the Bregma ±2.30 and ±3.80, Plate 29–34, according to Paxinos & Wattson Atlas). The sections were incubated for 48 h at 4°C with polyclonal rabbit anti-GFAP antiserum (Dako, UK, 1:500) and polyclonal mouse anti-NeuN (Chemicon, 1:250) in tris buffered (pH 7.4) saline anti-serum (TBS) containing 0.2% of Triton-X 100 and 10% of bovine serum. After being washed several times with TBS, the sections were incubated with 594 alexa-conjugated goat antirabbit and 488 alexa-conjugated rabbit antimouse antibodies for 2 h at room temperature. The sections were mounted on slides coated with 2% gelatin with chromium and potassium sulfate, using Vectashield mounting medium (Vector Laboratories, São Paulo, Brazil). All sections were photographed with a confocal microscope (Olympus, Japan).
Astrocytic immunohistochemical analyzes
The number of GFAP-stained astrocytes/mm2 in the hippocampus—CA1 was estimated according to previously reported [49]. All lighting conditions and magnifications were kept constant. The images were captured and a square region of interest (ROI) was created considering the minor size of the organ/layer. The ROI square was overlaid in all sections of stratum radiatum of CA1, with blood vessels and artifacts being avoided. Eight images were analyzed per field from a total of five animals per group. GFAP-reactive astrocytes located inside this square or intersected by the lower and/or right edges of the square were counted. Astrocytes intersected by the upper and/or left edges of the square were not counted [50]. Morphological analysis was performed by three separate observers who were blind to the experimental groups, and results were averaged in the final results.
Statistical analysis
The data obtained are expressed as mean ± SEM of mean. As the variances of the data were homogenous (analyzed by the Kolmogorov–Smirnov test), behavioral performance in reference memory task was analyzed by two-way repeated-measures analysis of variance (ANOVA) followed by the Bonferroni posttest when indicated. Mean escape latency was the dependent variable, day was the within-subjects variable, and the four groups were the between-subjects variables. Remaining data were analyzed by two-way ANOVA (factors were surgery and treatment). In all statistical comparisons, P < 0.05 was used as the criterion for significance.
Results
Behavioral and treatment parameters
Chronic cerebral hypoperfusion had no effect on liquid and food consumption or body weight during 6 months (data not shown). In addition, ad libitum 0.5 mg/mL GUO oral treatment did not affect the parameters mentioned above.
Purine CSF and plasma level
As shown in Table 1a, 6 weeks after chronic cerebral hypoperfusion, the 2VO-CT animals presented a significant higher UA and a lower INO CSF level compared to SHAM-CT animals (P < 0.05). No other purine level significantly differed between these two groups. In addition, 6 weeks of orally chronic GUO administration significant increased the CSF level of XAN compared to control groups (P < 0.05) but did not significantly changed the GUO nor another CSF purine level investigated here.
As shown in Table 1b, the plasma level of several purines investigated were changed after 6 weeks of chronic cerebral hypoperfusion. The levels of GMP, XAN, UA and ADO were significant higher in the plasma of 2VO-CT compared to SHAM-CT animals (P < 0.05). Regarding GUO treatment, GUO administration significantly increased XAN and ADO levels while decreased HIPOX level compared to SHAM-CT (P < 0.05). In addition, Two Way ANOVA of variances analysis pointed that there was interaction between the surgery and the treatment on plasma GUO and ADO levels [F(1.28) = 5.06, P = 0.03].
Morris water maze learning
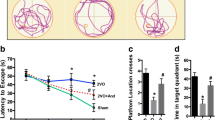
All rats showed similar escape latencies on training day 1, a two-way ANOVA with one between factor (4 groups) versus five repeated measures (escape latencies) indicated a significant difference among groups [F(3, 144) = 6.36, P < 0.05], and a significant difference over training days [F(4, 133) = 7.64, P < 0.01] (Fig. 1a). The escape latency of both sham groups (CT and GUO) decreased from the 1st to the 5th day of the task, while both 2VO groups (CT and GUO) did not present this performance, indicating that 2VO procedure impaired the learning of this task and that GUO did not interfere with this impairment.

Performance of rats in the water maze task submitted to chronic cerebral hypoperfusion evaluated. a Curves in the Morris water maze performance in the reference memory protocol. 2VO groups showed significantly longer escape latencies than SHAM groups, respectively (*P < 0.05, # P < 0.05). b Memory time spent in the target quadrant showing memory impairment of 2VO-CT and 2VO-GUO compared to SHAM-CT and SHAM-GUO groups (*P < 0.05). N = 10 animals per group
In the probe test, both sham groups (CT and GUO) spent more time in the former platform quadrant (Fig. 1b) than both 2VO groups (CT and GUO) [by two-way ANOVA, F(1, 35) = 5.20, P < 0.01]. Again, GUO had no effect on this performance. In addition, it was not observed any motor deficit: the mean swimming speed was 0.15 m/s for control group and 0.17 m/s for 2VO group.
Brain insult markers
As shown in Fig. 2, CSF BDNF (A) and serum S100B (B) levels were not affected by 2VO procedure and/or by GUO administration, by two way ANOVA (P > 0.05).

CSF BDNF and serum S100B levels. No differences were observed in CSF BDNF concentration (a) and in serum S100B (b) (P > 0.05). N = 10 animals per group
Hippocampal damage
Immunohistochemistry was carried out using NeuN and GFAP for labeling neurons and astrocytes, respectively. The hippocampal damage was evaluated 6 months after 2VO-surgery. Evident loss of CA1 pyramidal neurons and increased reactive glial labeling cells was observed in 2 of 10 animals in the 2VO-CT group (Fig. 3). No difference was detected in the NeuN or GFAP labeling in CA3 or DG (Dentate Gyrus) regions. Representative images of hippocampal CA1, CA3 and DG from rats of the four groups are shown in Figs. 3, 4 and 5, respectively.

Pyramidal neurons and GFAP immunoreactivity in the CA1 hippocampal region of 2VO-CT rats six month after chronic cerebral hypoperfusion. Immunofluorescence showing NeuN (a, d, g, j) and GFAP (b, e, h, k) positive cells in SHAM-CT (a–c), SHAM-GUO (d–f), 2VO-CT (g–i) and 2VO-GUO (j–l) groups. ×20, scale bar 100 μm

Pyramidal neurons and GFAP immunoreactivity in the CA3 hippocampal region of rats. Immunofluorescence showing NeuN (a, d, g, j) and GFAP (b, e, h, k) positive cells in SHAM-CT (a–c), SHAM-GUO (d–f), 2VO-CT (g–i) and 2VO-GUO (j–l) groups. ×20, scale bar 100 μm

Pyramidal neurons and GFAP immunoreactivity in the DG hippocampal region. Immunofluorescence showing NeuN (a, d, g, j) and GFAP (b, e, h, k) positive cells in SHAM-CT (a–c), SHAM-GUO (d–f), 2VO-CT (g–i) and 2VO-GUO (j–l) groups. ×20, scale bar 100 μm
Astrocytic cell bodies and processes were identified in the CA1 stratum radiatum of all animals excepting the two rats displaying pronounced reactive glial labeling. There was no significant difference in the number and/or in the morphology of astrocytes among the groups (data not shown).
Discussion
The results of the present study demonstrate that chronic cerebral hypoperfusion produced by 2VO, impaired spatial learning and memory evaluated by Morris water maze task and caused severe hippocampal damage in 20% of the animals. The behavioral effect was not affected by GUO treatment. However, no hippocampal damage was observed in the 2VO animals treated with GUO. In addition, no alteration was observed in neither the CSF BDNF levels nor in the serum S100B concentration caused by 2VO surgery and/or by GUO treatment.
Studies involving the use of the 2VO model have demonstrated that an impaired spatial learning function is associated with hippocampal damage [6, 10, 12]. In our study, we did not observe this correlation: animals presenting behavioral alteration did not present cell hippocampal damage. We cannot exclude that other cell damage not investigated here may be present in the 2VO group. In fact, the relationship between chronic cerebral hypoperfusion produced by 2VO and impaired cognitive function has not been completely elucidated. Other studies also found almost no correlation between the loss of CA1 neurons and the Morris maze performance [51–53]. The pyramidal and granular cells in the hippocampus are not the exclusive cells involved in spatial memory. Other regions of the hippocampal formation such as the entorhinal cortex, the parahippocampal gyrus, and the rhinal and cingular gyri are also involved in spatial memory [54, 55]. Moreover, the memory deficit correlates with the white matter damage in 2VO rats [6, 56–58]. Thus, a direct link cannot be established between 2VO-induced memory failure and a specific appearance of neuronal damage in the hippocampus.
It is noteworthy that variable hippocampal damage has been reported by others [6, 59–61]. As in most vascular models, individual anatomical differences in the cerebrovascular anatomy at the circle of Willis of rodents can influence experimental variability [62]. Differences in the focal lesions outcome after 2VO in different laboratories may be explained possibly by differences in rat strain, animal age at the time of occlusion or anesthesia.
The hippocampal damage here observed in two animals was evidenced by a massive neuronal loss in the CA1 region that was accompanied by an intense increase in GFAP labeling, indicating an astrogliosis process. No alteration in the CA3 or DG areas was observed. This is consistent with the selective vulnerability of CA1 neurons that has been observed in others studies using the 2VO model and with various acute ischemia models [6, 8, 63]. GFAP is commonly used as a marker for changes in astroglial cells during brain development and injury [64]. In fact, CNS injury, as consequence of brain diseases as trauma, ischemia, genetic disorders, neurodegenerative disorders or chemical insult causes astrocytes to become reactive, a condition accompanied by an increase in GFAP levels [65].
In our study, 2VO procedure did not affect the CSF BDNF level and the serum S100B concentration in rats. BDNF and S100B are two proteins commonly used as biomarkers for brain diseases, including vascular and Alzheimer’s diseases [66–73]. BDNF in the hippocampus is critical for the acquisition and/or consolidation of spatial memory [74–76]. Previous studies showed that chronic cerebral hypoperfusion induced down-regulation of hippocampal BDNF [77]. In addition, 2VO rats presenting an increase in hippocampal BDNF levels also presented better performance in the water maze task [77]. S100B is a calcium-binding protein found in brain tissue, predominantly in astrocytes. This protein has putative intra and extracellular functions. Intracellular roles include regulation of protein phosphorylation, cytoskeleton components and transcriptional factors [78, 79]; extracellularly, S100B plays trophic roles on neuronal and glial cells, but elevated levels of this protein could induce apoptosis in neural cells [80]. In a previous work, we demonstrated that the hippocampal S100B levels increased, while CSF S100B levels decreased, after the 2VO surgery [48]. To note, previous works evaluated the BDNF and S100B proteins 8 and 10 weeks after the 2VO surgery, respectively [48, 77]. In the present work, we investigated the 2VO effects 6 months after the surgery. Therefore, we cannot rule out that alteration in the CSF BDNF level or serum S100B concentration may be altered in other periods after 2VO surgery.
Glutamatergic excitotoxicity is suggested to play a pivotal role in the behavioral alterations and neuronal damage observed in ischemic insult, including hypoperfusion [19, 20, 48]. In a previous work, we demonstrated that 2VO animals presented a decrease of hippocampal glutamate uptake, indicating a higher susceptibility of these animals to excitotoxicity [48]. A correlation between hippocampal glutamate uptake and Morris water maze performance was also evidenced [48]. Our group has demonstrated that acute in vitro administration of GUO stimulates glutamate uptake [40, 41, 44], and in vivo administration prevents the glutamate uptake decrease after excitotoxic stimuli [38, 39, 81, 82]. Therefore, we expected that GUO treatment would be able to prevent the behavioral alterations observed in 2VO rats, which did not occur in this study. Of note, none of the animals that received GUO treatment after the 2VO surgery showed neuronal injury.
GUO was given orally for 6 weeks immediately from the 2VO surgery. We chose to treat the animals with chronic orally treatment because we previously demonstrated that GUO chronic administration was neuroprotective against excitotoxicity insults [37, 82]. However, no other study, to our knowledge, administrated GUO for such a long period as 6 weeks as we did it here. Surprising, we could not observe any difference in the CSF or plasma GUO level after the treatment (Table 1). Of note, we observed that the level of XAN (one direct product of GUO degradation) is increased at CSF and plasma. Additionally, GUO administration increased the plasma ADO and decreased HIPOX levels, suggesting some change in the homeostasis of the purinergic system. Importantly, previous works already demonstrated that GUO can be metabolized both in plasma [83] and CSF [84]. As we already have demonstrated that 2 weeks of orally chronic GUO treatment increased CSF GUO levels [82], we can speculate that the long period of GUO treatment used here (6 weeks) could, at somehow, accelerate GUO degradation. In fact, it is important to mention that both the CSF and plasma levels of most of the purines evaluated here showed a higher variability between the GUO treated rats in contrast to the control animals. In this way, other protocols using GUO treatment with different administration schedule should be design to investigate the GUO effects in 2VO animals.
Noteworthy, the plasma level of several purines (GMP, XAN, UA and ADO) were higher after 6 weeks of chronic cerebral hypoperfusion compared to control animals (Table 1b). Regarding CSF, only UA level was higher and INO level was lower in the 2VO-CT animals compared to SHAM-CT group. Although the meanings of these results are difficult to explain here, it could suggest that the purinergic system could play an important role in the pathophysiology of chronic cerebral hypopefusion.
In summary, we did not observe correlation between the cognitive impairment and hippocampal cell damage in the 2VO animals. In addition, CSF BDNF and serum S100B levels were not brain injury markers, at least 6 months after the 2VO surgery. Although the GUO neuroprotective potential in experimental models involving excitotoxicity events has been well demonstrated, in the present work GUO chronic orally treatment did not prevent the behavioral alterations observed in 2VO animals. We are continuing to characterize the GUO effects on the behavioral and histological alterations observed by chronic cerebral hypoperfusion.
References
Desmond DW (2004) The neuropsychology of vascular cognitive impairment: is there a specific cognitive deficit? J Neurol Sci 226(1–2):3–7. doi:10.1016/j.jns.2004.09.002
Rockwood K, Wentzel C, Hachinski V, Hogan DB, MacKnight C, McDowell I (2000) Prevalence and outcomes of vascular cognitive impairment. Vascular Cognitive Impairment Investigators of the Canadian Study of Health and Aging. Neurology 54(2):447–451
Farkas E, Luiten PG (2001) Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 64(6):575–611. pii:S0301-0082(00)00068-X
Ohnishi T, Matsuda H, Tabira T, Asada T, Uno M (2001) Changes in brain morphology in Alzheimer disease and normal aging: is Alzheimer disease an exaggerated aging process? AJNR Am J Neuroradiol 22(9):1680–1685
Borroni B, Di Luca M, Padovani A (2006) Predicting Alzheimer dementia in mild cognitive impairment patients. Are biomarkers useful? Eur J Pharmacol 545(1):73–80. doi:10.1016/j.ejphar.2006.06.023
Farkas E, Luiten PG, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54(1):162–180. doi:10.1016/j.brainresrev.2007.01.003
Marshall RS, Lazar RM, Pile-Spellman J, Young WL, Duong DH, Joshi S, Ostapkovich N (2001) Recovery of brain function during induced cerebral hypoperfusion. Brain 124(Pt 6):1208–1217
Ni J, Ohta H, Matsumoto K, Watanabe H (1994) Progressive cognitive impairment following chronic cerebral hypoperfusion induced by permanent occlusion of bilateral carotid arteries in rats. Brain Res 653(1–2):231–236
Ni JW, Matsumoto K, Li HB, Murakami Y, Watanabe H (1995) Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res 673(2):290–296. 0006-8993(94)01436-L
Sarti C, Pantoni L, Bartolini L, Inzitari D (2002) Cognitive impairment and chronic cerebral hypoperfusion: what can be learned from experimental models. J Neurol Sci 203–204:263–266. pii:S0022510X02003027
Briede J, Duburs G (2007) Protective effect of cerebrocrast on rat brain ischaemia induced by occlusion of both common carotid arteries. Cell Biochem Funct 25(2):203–210. doi:10.1002/cbf.1318
Otori T, Katsumata T, Muramatsu H, Kashiwagi F, Katayama Y, Terashi A (2003) Long-term measurement of cerebral blood flow and metabolism in a rat chronic hypoperfusion model. Clin Exp Pharmacol Physiol 30(4):266–272. 3825
Tsuchiya M, Sako K, Yura S, Yonemasu Y (1993) Local cerebral glucose utilisation following acute and chronic bilateral carotid artery ligation in Wistar rats: relation to changes in local cerebral blood flow. Exp Brain Res 95(1):1–7
Chao XD, Fei F, Fei Z (2010) The role of excitatory amino acid transporters in cerebral ischemia. Neurochem Res 35(8):1224–1230. doi:10.1007/s11064-010-0178-3
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397. pii:S0166-2236(99)01401-0
Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 460(2):525–542. doi:10.1007/s00424-010-0809-1
Segovia G, Porras A, Del Arco A, Mora F (2001) Glutamatergic neurotransmission in aging: a critical perspective. Mech Ageing Dev 122(1):1–29. pii:S0047-6374(00)00225-6
Szydlowska K, Tymianski M (2010) Calcium, ischemia and excitotoxicity. Cell Calcium 47(2):122–129. doi:10.1016/j.ceca.2010.01.003
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30(4):379–387. doi:10.1038/aps.2009.24
Marosi M, Fuzik J, Nagy D, Rakos G, Kis Z, Vecsei L, Toldi J, Ruban-Matuzani A, Teichberg VI, Farkas T (2009) Oxaloacetate restores the long-term potentiation impaired in rat hippocampus CA1 region by 2-vessel occlusion. Eur J Pharmacol 604(1–3):51–57. doi:10.1016/j.ejphar.2008.12.022
Bau C, Middlemiss PJ, Hindley S, Jiang S, Ciccarelli R, Caciagli F, Diiorio P, Werstiuk ES, Rathbone MP (2005) Guanosine stimulates neurite outgrowth in PC12 cells via activation of heme oxygenase and cyclic GMP. Purinergic Signal 1(2):161–172. doi:10.1007/s11302-005-6214-0
Ciccarelli R, Di Iorio P, D’Alimonte I, Giuliani P, Florio T, Caciagli F, Middlemiss PJ, Rathbone MP (2000) Cultured astrocyte proliferation induced by extracellular guanosine involves endogenous adenosine and is raised by the co-presence of microglia. Glia 29(3):202–211. doi:10.1002/(SICI)1098-1136(20000201)29:3<202:AID-GLIA2>3.0.CO;2-C
Ciccarelli R, Ballerini P, Sabatino G, Rathbone MP, D’Onofrio M, Caciagli F, Di Iorio P (2001) Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci 19(4):395–414. pii:S0736574800000848
Kim JK, Rathbone MP, Middlemiss PJ, Hughes DW, Smith RW (1991) Purinergic stimulation of astroblast proliferation: guanosine and its nucleotides stimulate cell division in chick astroblasts. J Neurosci Res 28(3):442–455. doi:10.1002/jnr.490280318
Middlemiss PJ, Gysbers JW, Rathbone MP (1995) Extracellular guanosine and guanosine-5′-triphosphate increase: NGF synthesis and release from cultured mouse neopallial astrocytes. Brain Res 677(1):152–156. pii:0006-8993(95)00156-K
Rathbone M, Pilutti L, Caciagli F, Jiang S (2008) Neurotrophic effects of extracellular guanosine. Nucleosides Nucleotides Nucleic Acids 27(6):666–672. doi:10.1080/15257770802143913
Schmidt AP, Lara DR, Souza DO (2007) Proposal of a guanine-based purinergic system in the mammalian central nervous system. Pharmacol Ther 116(3):401–416. doi:10.1016/j.pharmthera.2007.07.004
Schmidt AP, Paniz L, Schallenberger C, Bohmer AE, Wofchuk ST, Elisabetsky E, Portela LV, Souza DO (2009) Guanosine prevents thermal hyperalgesia in a rat model of peripheral mononeuropathy. J Pain. doi:10.1016/j.jpain.2009.06.010
Schmidt AP, Souza DO (2010) The role of the guanine-based purinergic system in seizures and epilepsy. Open Neurosci J 4:102–113. doi:10.2174/1874082001004010113
Lara DR, Schmidt AP, Frizzo ME, Burgos JS, Ramirez G, Souza DO (2001) Effect of orally administered guanosine on seizures and death induced by glutamatergic agents. Brain Res 912(2):176–180. pii:S0006-8993(01)02734-2
Schmidt AP, Lara DR, de Faria Maraschin J, da Silveira Perla A, Onofre Souza D (2000) Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Res 864(1):40–43. pii:S0006-8993(00)02106-5
Schmidt AP, Avila TT, Souza DO (2005) Intracerebroventricular guanine-based purines protect against seizures induced by quinolinic acid in mice. Neurochem Res 30(1):69–73
Schmidt AP, Bohmer AE, Leke R, Schallenberger C, Antunes C, Pereira MS, Wofchuk ST, Elisabetsky E, Souza DO (2008) Antinociceptive effects of intracerebroventricular administration of guanine-based purines in mice: evidences for the mechanism of action. Brain Res 1234:50–58. doi:10.1016/j.brainres.2008.07.091
Schmidt AP, Bohmer AE, Antunes C, Schallenberger C, Porciuncula LO, Elisabetsky E, Lara DR, Souza DO (2009) Anti-nociceptive properties of the xanthine oxidase inhibitor allopurinol in mice: role of A1 adenosine receptors. Br J Pharmacol 156(1):163–172. doi:10.1111/j.1476-5381.2008.00025.x
Soares FA, Schmidt AP, Farina M, Frizzo ME, Tavares RG, Portela LV, Lara DR, Souza DO (2004) Anticonvulsant effect of GMP depends on its conversion to guanosine. Brain Res 1005(1–2):182–186. doi:10.1016/j.brainres.2004.01.053
Vinade ER, Izquierdo I, Lara DR, Schmidt AP, Souza DO (2004) Oral administration of guanosine impairs inhibitory avoidance performance in rats and mice. Neurobiol Learn Mem 81(2):137–143. doi:10.1016/j.nlm.2003.12.003
Vinade ER, Schmidt AP, Frizzo ME, Izquierdo I, Elisabetsky E, Souza DO (2003) Chronically administered guanosine is anticonvulsant, amnesic and anxiolytic in mice. Brain Res 977(1):97–102. pii:S0006899303027690
Moretto MB, Arteni NS, Lavinsky D, Netto CA, Rocha JB, Souza DO, Wofchuk S (2005) Hypoxic-ischemic insult decreases glutamate uptake by hippocampal slices from neonatal rats: prevention by guanosine. Exp Neurol 195(2):400–406. doi:10.1016/j.expneurol.2005.06.005
Moretto MB, Boff B, Lavinsky D, Netto CA, Rocha JB, Souza DO, Wofchuk ST (2009) Importance of schedule of administration in the therapeutic efficacy of guanosine: early intervention after injury enhances glutamate uptake in model of hypoxia-ischemia. J Mol Neurosci 38(2):216–219. doi:10.1007/s12031-008-9154-7
Frizzo ME, Antunes Soares FA, Dall’Onder LP, Lara DR, Swanson RA, Souza DO (2003) Extracellular conversion of guanine-based purines to guanosine specifically enhances astrocyte glutamate uptake. Brain Res 972(1–2):84–89. pii:S000689930302506X
Frizzo ME, Lara DR, Dahm KC, Prokopiuk AS, Swanson RA, Souza DO (2001) Activation of glutamate uptake by guanosine in primary astrocyte cultures. Neuroreport 12(4):879–881
Frizzo ME, Lara DR, Prokopiuk Ade S, Vargas CR, Salbego CG, Wajner M, Souza DO (2002) Guanosine enhances glutamate uptake in brain cortical slices at normal and excitotoxic conditions. Cell Mol Neurobiol 22(3):353–363
Frizzo ME, Schwalm FD, Frizzo JK, Soares FA, Souza DO (2005) Guanosine enhances glutamate transport capacity in brain cortical slices. Cell Mol Neurobiol 25(5):913–921. doi:10.1007/s10571-005-4939-5
Gottfried C, Tramontina F, Goncalves D, Goncalves CA, Moriguchi E, Dias RD, Wofchuk ST, Souza DO (2002) Glutamate uptake in cultured astrocytes depends on age: a study about the effect of guanosine and the sensitivity to oxidative stress induced by H2O2. Mech Ageing Dev 123(10):1333–1340. pii:S0047637402000696
Ohta H, Nishikawa H, Kimura H, Anayama H, Miyamoto M (1997) Chronic cerebral hypoperfusion by permanent internal carotid ligation produces learning impairment without brain damage in rats. Neuroscience 79(4):1039–1050. pii:S0306-4522(97)00037-7
Schmidt-Kastner R, Aguirre-Chen C, Saul I, Yick L, Hamasaki D, Busto R, Ginsberg MD (2005) Astrocytes react to oligemia in the forebrain induced by chronic bilateral common carotid artery occlusion in rats. Brain Res 1052(1):28–39. doi:10.1016/j.brainres.2005.06.018
Cechetti F, Worm PV, Pereira LO, Siqueira IR, A Netto C (2010) The modified 2VO ischemia protocol causes cognitive impairment similar to that induced by the standard method, but with a better survival rate. Braz J Med Biol Res 43(12):1178–1183. pii:S0100-879X2010007500124
Vicente E, Degerone D, Bohn L, Scornavaca F, Pimentel A, Leite MC, Swarowsky A, Rodrigues L, Nardin P, de Almeida LM, Gottfried C, Souza DO, Netto CA, Goncalves CA (2009) Astroglial and cognitive effects of chronic cerebral hypoperfusion in the rat. Brain Res 1251:204–212. doi:10.1016/j.brainres.2008.11.032
Martinez FG, Hermel EE, Xavier LL, Viola GG, Riboldi J, Rasia-Filho AA, Achaval M (2006) Gonadal hormone regulation of glial fibrillary acidic protein immunoreactivity in the medial amygdala subnuclei across the estrous cycle and in castrated and treated female rats. Brain Res 1108(1):117–126. doi:10.1016/j.brainres.2006.06.014
Viola GG, Rodrigues L, Americo JC, Hansel G, Vargas RS, Biasibetti R, Swarowsky A, Goncalves CA, Xavier LL, Achaval M, Souza DO, Amaral OB (2009) Morphological changes in hippocampal astrocytes induced by environmental enrichment in mice. Brain Res 1274:47–54. doi:10.1016/j.brainres.2009.04.007
Institoris A, Farkas E, Berczi S, Sule Z, Bari F (2007) Effects of cyclooxygenase (COX) inhibition on memory impairment and hippocampal damage in the early period of cerebral hypoperfusion in rats. Eur J Pharmacol 574(1):29–38. doi:10.1016/j.ejphar.2007.07.019
Jaspers RM, Block F, Heim C, Sontag KH (1990) Spatial learning is affected by transient occlusion of common carotid arteries (2VO): comparison of behavioural and histopathological changes after ‘2VO’ and ‘four-vessel-occlusion’ in rats. Neurosci Lett 117(1–2):149–153
Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, Young HF, Hayes RL (1990) Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res 526(2):249–258. pii:0006-8993(90)91229-A
Meunier M, Hadfield W, Bachevalier J, Murray EA (1996) Effects of rhinal cortex lesions combined with hippocampectomy on visual recognition memory in rhesus monkeys. J Neurophysiol 75(3):1190–1205
Wiig KA, Bilkey DK (1994) The effects of perirhinal cortical lesions on spatial reference memory in the rat. Behav Brain Res 63(1):101–109. pii:0166-4328(94)90055-8
Wakita H, Tomimoto H, Akiguchi I, Kimura J (1994) Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunohistochemical study. Acta Neuropathol 87(5):484–492
Lee JH, Park SY, Shin YW, Hong KW, Kim CD, Sung SM, Kim KY, Lee WS (2006) Neuroprotection by cilostazol, a phosphodiesterase type 3 inhibitor, against apoptotic white matter changes in rat after chronic cerebral hypoperfusion. Brain Res 1082(1):182–191. doi:10.1016/j.brainres.2006.01.088
Watanabe T, Zhang N, Liu M, Tanaka R, Mizuno Y, Urabe T (2006) Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke 37(6):1539–1545. doi:10.1161/01.STR.0000221783.08037.a9
Bennett SA, Tenniswood M, Chen JH, Davidson CM, Keyes MT, Fortin T, Pappas BA (1998) Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. Neuroreport 9(1):161–166
Pappas BA, de la Torre JC, Davidson CM, Keyes MT, Fortin T (1996) Chronic reduction of cerebral blood flow in the adult rat: late-emerging CA1 cell loss and memory dysfunction. Brain Res 708(1–2):50–58. pii:0006-8993(95)01267-2
Plaschke K, Grant M, Weigand MA, Zuchner J, Martin E, Bardenheuer HJ (2001) Neuromodulatory effect of propentofylline on rat brain under acute and long-term hypoperfusion. Br J Pharmacol 133(1):107–116. doi:10.1038/sj.bjp.0704061
Schmidt-Kastner R, Eysel UT (1994) Ischemic damage visualized in flat mounts of rat retina after photochemically induced thrombosis. Brain Res Bull 34(5):487–491. pii:0361-9230(94)90022-1
Bowler JV (2004) Vascular cognitive impairment. Stroke 35(2):386–388. doi:10.1161/01.STR.0000115301.12426.2B35/2/386
Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25(9–10):1439–1451
O’Callaghan JP, Sriram K (2005) Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf 4(3):433–442. doi:10.1517/14740338.4.3.433
Chaves ML, Camozzato AL, Ferreira ED, Piazenski I, Kochhann R, Dall’Igna O, Mazzini GS, Souza DO, Portela LV (2010) Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J Neuroinflamm 7:6. doi:10.1186/1742-2094-7-6
Grillo RW, Ottoni GL, Leke R, Souza DO, Portela LV, Lara DR (2007) Reduced serum BDNF levels in schizophrenic patients on clozapine or typical antipsychotics. J Psychiatr Res 41(1–2):31–35. doi:10.1016/j.jpsychires.2006.01.005
Haas L, Portela LV, Bohmer AE, Oses JP, Lara DR (2010) Increased plasma levels of brain derived neurotrophic factor (BDNF) in patients with fibromyalgia. Neurochem Res 35(5):830–834. doi:10.1007/s11064-010-0129-z
Kessler FH, Woody G, Portela LV, Tort AB, De Boni R, Peuker AC, Genro V, von Diemen L, de Souza DO, Pechansky F (2007) Brain injury markers (S100B and NSE) in chronic cocaine dependents. Rev Bras Psiquiatr 29(2):134–139
Lee J, Fukumoto H, Orne J, Klucken J, Raju S, Vanderburg CR, Irizarry MC, Hyman BT, Ingelsson M (2005) Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp Neurol 194(1):91–96. doi:10.1016/j.expneurol.2005.01.026
Machado-Vieira R, Dietrich MO, Leke R, Cereser VH, Zanatto V, Kapczinski F, Souza DO, Portela LV, Gentil V (2007) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry 61(2):142–144. doi:10.1016/j.biopsych.2006.03.070
Schaf DV, Tort AB, Fricke D, Schestatsky P, Portela LV, Souza DO, Rieder CR (2005) S100B and NSE serum levels in patients with Parkinson’s disease. Parkinsonism Relat Disord 11(1):39–43. doi:10.1016/j.parkreldis.2004.07.002
Zhang J, Sokal I, Peskind ER, Quinn JF, Jankovic J, Kenney C, Chung KA, Millard SP, Nutt JG, Montine TJ (2008) CSF multianalyte profile distinguishes Alzheimer and Parkinson diseases. Am J Clin Pathol 129(4):526–529. doi:10.1309/W01Y0B808EMEH12L
Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH (2002) BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus 12(4):551–560. doi:10.1002/hipo.10035
Alonso M, Vianna MR, Izquierdo I, Medina JH (2002) Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol 22(5–6):663–674
Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH (2007) Persistence of long-term memory storage requires a late protein synthesis- and BDNF-dependent phase in the hippocampus. Neuron 53(2):261–277. doi:10.1016/j.neuron.2006.11.025
Zheng P, Zhang J, Liu H, Xu X, Zhang X (2008) Angelica injection reduces cognitive impairment during chronic cerebral hypoperfusion through brain-derived neurotrophic factor and nerve growth factor. Curr Neurovasc Res 5(1):13–20
Donato R (2001) S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 33(7):637–668. pii:S1357-2725(01)00046-2
Goncalves CA, Leite MC, Nardin P (2008) Biological and methodological features of the measurement of S100B, a putative marker of brain injury. Clin Biochem 41(10–11):755–763. doi:10.1016/j.clinbiochem.2008.04.003
Van Eldik LJ, Wainwright MS (2003) The Janus face of glial-derived S100B: beneficial and detrimental functions in the brain. Restor Neurol Neurosci 21(3–4):97–108
de Oliveira DL, Horn JF, Rodrigues JM, Frizzo ME, Moriguchi E, Souza DO, Wofchuk S (2004) Quinolinic acid promotes seizures and decreases glutamate uptake in young rats: reversal by orally administered guanosine. Brain Res 1018(1):48–54. doi:10.1016/j.brainres.2004.05.033
Vinade ER, Schmidt AP, Frizzo ME, Portela LV, Soares FA, Schwalm FD, Elisabetsky E, Izquierdo I, Souza DO (2005) Effects of chronic administered guanosine on behavioral parameters and brain glutamate uptake in rats. J Neurosci Res 79(1–2):248–253. doi:10.1002/jnr.20327
Jiang S, Fischione G, Giuliani P, Romano S, Caciagli F, Di Iorio P (2008) Metabolism and distribution of guanosine given intraperitoneally: implications for spinal cord injury. Nucleosides Nucleotides Nucleic Acids 27(6):673–680. doi:10.1080/15257770802143962
Silva RG, Santos DS, Basso LA, Oses JP, Wofchuk S, Portela LV, Souza DO (2004) Purine nucleoside phosphorylase activity in rat cerebrospinal fluid. Neurochem Res 29(10):1831–1835
Acknowledgments
The authors are grateful to Rafael Berger Faraco, Vinicius Fornari Fernandes, Jocemar Ilha and Henrique Beck Biehl (laboratory technician) for their help in carrying out some of the experiments and Maria Elisa Calcagnotto for fruitful discussion. We also like to thank the Centro de Microscopia Eletrônica Confocal—UFRGS—(The Center of Confocal Microscopy—UFRGS) for the help with the microscopy technique used in this article. This research was supported by the Brazilian funding agencies FINEP “Rede Instituto Brasileiro de Neurociência (IBN-Net) # 01.06.08.42-00, CNPq, CAPES, FAPERGS, UFRGS and INCT for Excitotoxicity and Neuroprotection/CNPQ.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ganzella, M., de Oliveira, E.D.A., Comassetto, D.D. et al. Effects of chronic guanosine treatment on hippocampal damage and cognitive impairment of rats submitted to chronic cerebral hypoperfusion. Neurol Sci 33, 985–997 (2012). https://doi.org/10.1007/s10072-011-0872-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-011-0872-1