
Abstract
In the present study, we evaluated the neuroprotection time window for nerve growth factor (NGF) after ischemia/reperfusion brain injury in rabbits as related to this anti-apoptosis mechanism. Male New Zealand rabbits were subjected to 2 h of middle cerebral artery occlusion (MCAO), followed by 70 h of reperfusion. NGF was administered after injury to evaluate the time window. Neurological deficits, infarct volume, neural cell apoptosis and expressions of caspase-3 and Bcl-2 were measured. Compared to saline-treated control, NGF treatment at 2, 3 and 5 h after MCAO significantly reduced infarct volume, neural cell apoptosis and expression of caspase-3 (P < 0.01), up-regulated the expression of Bcl-2 and improved functional recovery (P < 0.01). However, treatment at latter time points did not produce significant neuroprotection. Neuroprotection treatment with NGF provides an extended time window of up to 5 h after ischemia/reperfusion brain injury, in part by attenuating the apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is the third cause of death after heart disease and cancer and is the leading cause of severe disability in the developed countries. Thrombolysis by intravenous administration recombinant tissue plasminogen activator (r-tpA) is the only Food and Drug Administration (FDA)-approved treatment for acute stroke. However, fewer than 5% of stroke patients receive this therapy because of the narrow time window (<3 h) [1]. Furthermore, once reflow is established, reperfusion injury may limit the usefulness of intervention [2]. These observations suggest that there is a compelling need to develop treatments of ischemic stroke designed specifically to reduce neurologic deficits, for example, neuroprotection, which can target the vast majority of patients. Many neuroprotective drugs interrupting one or more of the pathway of ischemic cell death after stroke have been effective in reducing infarct volume in various animal models, but have failed to demonstrate effective in-phase III human studies. Several concomitant factors have likely contributed to these failures, including poor preclinical pharmacodynamic evaluations, especially narrow treatment windows [3]. Obviously, the development of preclinical testing must focus on these neuroprotective factors to improve opportunities for successful transitions from preclinical to clinical studies [4]. So, the therapeutic time window becomes an important parameter to study and evaluate the neuroprotective effect [5].
Nerve growth factor (NGF) is an evolutionarily conserved polypeptide neurotrophin, which plays a crucial role in the sympathetic and sensory nervous systems [6]. Recent studies have indicated that NGF can prevent the metabolic or excitotoxic injury of cultured hippocampal and cortical neurons [7, 8]. NGF has also been shown to stimulate neurite outgrowth in PC12 cells by activating either PI-3 K or PLC-γ [9, 10]. Furthermore, mitogen-activated protein kinase (MAPK), which follows the PLC-γ activation after the phosphorylation of Trk receptors by neurotrophins, is involved in the process of neuronal differentiation [11]. Exogenous NGF can protect cultured hippocampal neurons from excitotoxic damage induced by a high concentration of glutamate, hypoglycemia and iron-induced degeneration [7, 12–14]. In brain ischemia, NGF mRNA expression is up-regulated transiently and this seems to be protective for neurons [7, 13, 15]. Upregulation of NGF after injury to the central nervous system (CNS) is probably an endogenous protective mechanism to maintain neuronal survival in lesioned brain regions. Similarly, pretreatment with NGF or NGF administration in the early post-ischemic period decreases ischemia-induced brain infarction, brain edema, density of terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) (+) neurons, and immunoreactivity of extracellular signal-regulated kinase (ERK) and caspase-3 in the ischemic cortex [7, 8, 15–22], which suggests that NGF may reduce ischemic insults by attenuating apoptosis and/or necrosis. However, this ultra-early administration is not practical in the clinical setting. Thus, there is an additional concern with later administration of NGF and few studies have assessed the effects of NGF when administered after the onset of ischemia. Therefore, in the present study, we investigated the therapeutic time window and molecular mechanisms for neuroprotective effects of NGF when administrated after focal ischemia injury in rabbits.
Materials and methods
Animals
A total of 78 male New Zealand white rabbits weighing 2.5–3.0 kg were randomly divided into three groups: NGF-treated group (n = 60), saline-treated group (n = 12) and sham-operated group (n = 6). The rabbits were housed in separated cages and the room was kept at 24 ± 1°C temperature and 50–60% humidity, under a 12:12-h light/dark cycle and with access to food and water ad libitum. All experimental procedures were approved by the local animal care committee and carried out in accordance with the guidelines of the National Institutes of Health on animal care and the ethical guidelines for investigation of experimental pain in conscious animals. Anesthesia was induced with intravenous injection of 20 mg/kg pentobarbital sodium and, if necessary, maintained with a further 5 mg/kg. PE-50 polyethylene tubing was inserted into the left femoral artery for monitoring arterial blood gases, serum glucose and body temperature before, during and after operation. For pain relief, the surgical wounds were anesthetized in advance by using 2% lidocaine (0.1 ml).
Surgery
MCA occlusion
The model of intraluminal suture was used for induction of focal cerebral ischemia, as formerly described [22, 23]. Briefly, the left common carotid, internal carotid and external carotid arteries were exposed via a midline incision in the neck. Then the left common and external carotid arteries were ligated proximally (near the bifurcation) with 4-0 surgical sutures. A guide wire (RF SP26137 M, TERUMO, Japan) with a diameter of 0.53 mm was inserted into the internal carotid artery from the distal common carotid artery until the tip occluded the origin of the middle cerebral artery (MCA). The placement of guide wire in the MCA was confirmed by a contact X-ray. The guide wire was maintained in place for 2 h and then withdrawn to allow reperfusion. Magnetic resonance imaging (MRI) measurements were performed in all rabbits between 1.75 and 2 h after MCA occlusion (MCAO). Animals subjected to the same surgery without vascular occlusion served as sham-operated group (n = 6).
Study design
Rabbits received intracerebral microinjection of 50 μl NGF (16 μg/l) [22] at different time points, 2, 3, 5 or 8 h post-MCAO (n = 15 per time point) by the perifocal region route. Control animals received saline (n = 12) at 2 h after onset of MCAO. After 2 h of MCAO and 70 h of reperfusion, neurological deficits, infarct volume, neural cell apoptosis and expression levels of caspase-3 and Bcl-2 were measured.
MRI protocol
Diffusion weighted imaging (DWI) was conducted to document injury and to provide a guide for intercerebral injection and dissecting tissues from ischemic regions. All animals were imaged in a 1.5-T scanner (Toshiba Visart, Japan), with quadrature knee coil. DWI was performed using a spin-echo echo-planar imaging sequence [TR = 12,000 ms, TE = 108 ms, NA = 3, 2 different b values (b = 0 and b = 900 s/mm2), FOV = 16.5 cm × 16.5 cm, matrix = 96 × 96, 5 slices, slice thickness = 5 mm]. If hyperintensity was observed in the MCA territory, apparent diffusion coefficient (ADC) maps were constructed by acquiring a set of five images with increasing diffusion gradient amplitude. The ADC-based measurements of the size of the ischemic core and penumbra regions were performed as follows: the ranges of the lowest ADC values and ADC values in the matching contralateral anatomic regions were derived from the ADC maps such that the region with ADC values lower than the mean of the lowest ADC values plus 1 SD was referred to as the core [24]. The region with ADC values higher than the mean of the ADC values plus 1 SD in the core but lower than the mean of normal ADC values minus 2 SDs in that region was referred to as the ischemic penumbra [24].
Administration
After the MRI examination, animals were placed in a stereotaxic frame, and a burr hole (<2 mm) was made at the left parietal skull 3 mm posterior from the infraorbital margin and 10 mm lateral to the midline till the dura mater. Our qualitative preliminary observations and previous research [22] suggest that a single dose of 50 μl NGF (16 μg/l) immediately after reperfusion by the route of perifocal region has protective effects, which could decrease immunoreactivity of ERK and caspase-3 in the ischemic cortex and hippocampus. So NGF was dissolved in PBS to prepare 16 μg/l as described previously [22]. Single intracerebral microinjection of 50 μl NGF was conducted directly into the left ischemic penumbra cortex over a 10-min time period at a depth of 6 mm from the skull surface using a stereotactic micromanipulator. The needle was retained in place for 5 min after each injection. The same volume of saline served as treatment for the saline-treated group (control). After injection, the defect of the skull was covered with bone wax and then the skin was sutured.
Neurological evaluation
Animals were examined for neurological function 24 and 72 h after the onset of occlusion. The neurological findings were scored according to Purdy scoring method [25]. Each animal was examined for motor function (score = 4), consciousness (score = 4), head turning (score = 1), circling (score = 1) and hemianopsia (score = 1). The lowest was 2 points, suggesting no neurological impairment; the highest was 11 points, suggesting that animals lost consciousness or died.
TTC staining
For quantitative infarct volume, rabbits were killed at 72 h of MCAO with overdosed barbiturate. After the skulls were removed, the brain was rapidly taken and cooled in cold saline for 10 min. The brains were then coronally sectioned into five 5-mm thick sections. The brain slices were incubated for 30 min in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C, and fixed by immersing in a 4% buffered formalin solution. Infarct size was quantified by using an image-processing software package (Beihang University, Beijing, China). To compensate for the effect of brain edema, the infarct volume was measured by a commonly used indirect method [26] [formula: infarct volume = contralateral hemisphere volume-(ipsilateral hemisphere volume-infarct volume)].
TUNEL assay
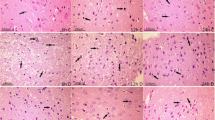
Neuronal damage was assessed by histological analysis of brain sections at 72 h of MCAO. The number of TUNEL-labeled cells in the penumbral cortex was counted on a computer screen grid from at least five random fields (×400) of each animal. Results were expressed as the average number of cells per field.
Flow cytometry analysis (percentage of apoptotic cells)
Flow cytometry was used to determine the extent of DNA fragmentation in cells extracted from the penumbral cortex. In separate rabbits (n = 5 per NGF-treated group, n = 4 saline-treated group, n = 2 sham-operated group), cortical tissue from the penumbral cortex was dissected immediately after killing. The cells in this tissue were dissociated by repeated aspiration in saline using a glass micropipette. Then a flow cytometer was used to calculate the cell apoptotic rate as reported by others [22, 27]. Absorbance ratio was measured at 488 nm of wavelength.
Immunohistochemistry
For histological examination, a total of 26 male New Zealand white rabbits (n = 5 per NGF-treated group, n = 4 saline-treated group, n = 2 sham-operated group) received pentobarbital sodium anesthesia and then were transcardially perfused with phosphate-buffered 4% paraformaldehyde. Five coronal paraffin sections (6 μm thick each) per animal corresponding to the same places in MR images were cut and processed for immunohistochemistry. The sections were washed in PBS, incubated in 3% H2O2 in PBS for 10 min, in blocking solution (10% goat serum in PBS) for 30 min at room temperature, and with rabbit polyclonal anti-caspase-3 (SC-7272) and anti-Bcl-2 (SC-7382, Wuhan Boshide Inc. Wuhan, China, 1:75) at 4°C overnight. Sections were washed and incubated. The reaction was stopped with DAB. The mean optical density values of the positive expression level of caspase-3 and Bcl-2 of five non-replicated fields were measured with the Meta Morph microimage analysis system under 400-fold light microscope.
Statistical analysis
All measurements in this study were performed blindly. Results were expressed as mean ± SD. Histological outcome measures were compared using one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons. Nonparametric ANOVA on ranks was used to compare total neurological scores among groups. Results were considered significant at P < 0.05.
Results
Infarct volume
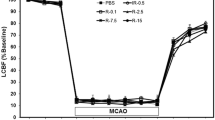
A total of 26 rabbits (n = 5 per NGF-treated group, n = 4 saline-treated group, n = 2 sham-operated group) were used for TTC analysis. No cerebral infarct was seen in the sham-operated rabbits. At 72 h of MCAO, a significant reduction of infarct volume was found when NGF was administered at 2, 3 or 5 h after MCAO, compared to the saline-treated group (P < 0.01, Fig. 1a). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (P > 0.05, Fig. 1a).

Quantitative effects of NGF on infarct volume (a) and neurological function (b) after MCAO (Mean ± SD). a NGF treatment 2, 3 and 5 h post-MCAO produced significant reduction in infarct volume as compared to saline-treated control at 24 or 72 h of MCAO. There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control. b NGF treatment 2, 3 and 5 h post-MCAO significantly improved neurological recovery as compared to saline-treated control at 24 or 72 h of MCAO. There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control; *versus MCAO group, P < 0.01
Neurological deficits
No neurological deficit was seen in sham-operated rabbits. When tested at 24 or 72 h of MCAO, saline-treated rabbits displayed a severe neurological deficit. NGF treatment 2, 3 and 5 h post-MCAO produced significant improvement in neurological score as compared to saline-treated control (Fig. 1b, P < 0.01). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (Fig. 1b, P > 0.05).
Flow cytometry analysis showing percentage of apoptotic cells
A total of 26 rabbits (n = 5 per NGF-treated group, n = 4 saline-treated group, n = 2 sham-operated group) were used for flow cytometric analysis. Cell apoptosis was characterised by a diploid nucleoliform peak (AP peak). Quantitative analysis of cell apoptosis could be performed according to the scale for which AP peak accounted. Apoptosis was rare in sham-operated group. At 72 h of MCAO, the percentage of apoptotic cells in the penumbral cortex markedly increased in saline-treated group than that in sham-operated group (P < 0.01, Fig. 2). The percentage of apoptotic cells in the penumbral cortex significantly reduced in NGF treatment 2, 3, and 5 h post-MCAO than in saline-treated group (P < 0.01, Fig. 2). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (P > 0.05, Fig. 2).

Flow cytometry analysis showing percentage of apoptotic cells at 72 h of MCAO. Apoptosis was rare in the sham-operated group (a). The percentage of apoptotic cells in the penumbral cortex was significantly reduced with NGF treatment 2 (b), 3, and 5 h post-MCAO than that in the saline-treated group. There was no significant difference between treatment with NGF 8 h (c) post-MCAO and saline-treated control. d Quantification of the percentage of apoptotic cells in the penumbral cortex; *versus saline-treated group (control), P < 0.01. # versus Sham-operated group, P < 0.01
TUNEL-positive cells
A total of 26 rabbits (n = 5 per NGF-treated group, n = 4 saline-treated group, n = 2 sham-operated group) were used for TUNEL staining. The number of TUNEL positive was rare in the sham-operated group. At 2 h of MCAO and 70 h of reperfusion, the number of TUNEL-positive cells in the penumbral cortex significantly decreased after treatment with NGF 2, 3 and 5 h post-MCAO compared to the saline-treated group (7.6 ± 1.5, 11.0 ± 2.9, 17.8 ± 2.4 vs. 32.8 ± 2.6, both P < 0.01). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (31.0 ± 2.9 vs. 32.8 ± 2.6, P > 0.05).
The expressions of caspase-3 and Bcl-2
In the sham group, weak caspase-3 immunoreactivity was detected. The expression of caspase-3 in the penumbral cortex markedly increased in saline-treated rabbits. Treatment with NGF 2, 3 and 5 h post-MCAO produced significant reduction in expression of caspase-3 as compared to saline-treated control (P < 0.01, Fig. 3). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (P > 0.05, Fig. 3).

Immunohistochemical staining for caspase-3 in the penumbral cortex after 72 h of MCAO. The photomicrographs showed caspase-3 expression in the penumbral cortex of animals that received NGF at 2 h (a), 3 h (b), 5 h (c) and 8 h (d, e) after MCAO and saline (f). g Microphotograph of negative control staining. a–c and e–g Bar 20 μm. d Bar 2 μm. H, Quantification of caspase-3 expression in the penumbral cortex after 72 h of MCAO; *versus saline-treated group (control), P < 0.01. I infarct area, P peri-infarct area. Dotted lines demarcate the infarct and peri-infarct areas
In the sham group, no Bcl-2 immunoreactivity was detected in the cerebral cortex. The expression of Bcl-2 in the penumbral cortex markedly increased in saline-treated rabbits. Treatment with NGF 2, 3 and 5 h post-MCAO significantly increased expression of Bcl-2 as compared to saline-treated control (P < 0.01, Fig. 4). There was no significant difference between treatment with NGF 8 h post-MCAO and saline-treated control (P > 0.05, Fig. 4).

Immunohistochemical staining for Bcl-2 in penumbral cortex after 72 h of MCAO. The photomicrographs showed Bcl-2 expression in the penumbral cortex of animals receiving saline (c) and NGF 2 h (a) and 8 h (b) after MCAO. Bar 20 μm. d Quantification of Bcl-2 expression in penumbral cortex after 72 h of MCAO; *versus saline-treated group (control), P < 0.01
Discussion
The extent of the ischemic penumbra is critical to the success of any acute stroke therapy, including thrombolysis. Patients without a penumbra are unlikely to benefit from any acute tissue salvage therapies, as irreversible injury had already occurred [28]. Conversely, patients with penumbral tissue may benefit from tissue salvage therapies, regardless of the duration of symptoms [28]. A necessary prerequisite of the effective therapeutic strategy is the existence of functionally impaired but viable and potentially salvageable brain tissue (i.e., penumbra) [29].
NGF is the prototype of the neurotrophin family of growth factor molecules [30–32]. NGF regulates the growth, development and plasticity of selective neuronal populations in the nervous system [33–36]. It acts through binding and activating specific cell surface receptors named trkA and p-75 [37, 38]. Neuronal NGF expression in vivo is markedly up-regulated by seizures, forebrain ischemia, marked hypoglycemia and tissue injury [28, 39, 40]. NGF promotes the survival of specific groups of neurons, both in vivo and in vitro [15, 41–46], and has a protective effect against delayed neuronal death (DND) after cerebral ischemia [15, 47].
The therapeutic time window of an agent interests people mostly, because increasing research has suggested that many neuroprotective agents had a special therapeutic time window. The failure of clinical trials with a number of neuroprotective drugs, which had previously shown dramatic efficacy in animal ischemia models, was due to inadequate investigation of the therapeutic time window [48]. Therefore, it is necessary to establish the time window for efficacy in animal models as a part of the evaluation of neuroprotection.
In the present study, NGF produced significant neuroprotection in focal cerebral ischemia, as evident from significant reduction in cerebral infarction and neurological deficits. The neuroprotection was found to be maximal in 2 h post-MCAO treatment groups. NGF treatment even up to 5 h post-MCAO produced significant reduction in infarct volume and neurological deficits. Treatment at latter time point (8 h post-MCAO) did not produce significant neuroprotection. Thus, we concluded that the therapeutic time window for NGF in rabbit focal cerebral ischemia model should be limited within 5 h after ischemia.
However, it is difficult to estimate the therapeutic time window in stroke patients from the animal data, but we can reasonably speculate that NGF may possess a considerably longer time window for successful therapeutic intervention in the clinical setting, according to recent studies hat suggest that the therapeutic time window might be wider in human strokes than in animal models [48, 49]. This implied that NGF might be of clinical value for the treatment of acute stroke.
In discussing the limitations of these studies using NGF for stroke, it is important to consider the animal models used and the timing and routes of drug delivery [50]. Experimental data from transient focal cerebral ischemia indicate that delayed reperfusion contributes to ischemic injury and increases the size of the ischemic lesion [51, 52]. Other experiments showed that mortality increased to 45% if reperfusion was initiated 120 min after MCAO [53]. The pathophysiological mechanism of injury from delayed reperfusion itself is not yet fully understood, but may include breakdown of the blood–brain barrier, increase of postischemic edema, secondary increase of excitatory amino acids, inflammatory reaction, and altered microvascular permeability and integrity [53–55]. In our study, NGF reduced significantly cerebral infarct volume when it was induced directly after a 120-min MCAO. NGF plays a key role in neuroprotection of CNS against ischemic damage. But its access to the CNS is restricted by poor blood–brain barrier (BBB) permeability because of its large molecular weight [56]. Besides that, peripheral adverse effects following systemic administration also limit the clinical use of NGF [57, 58]. Though intracerebroventricular administration or grafting of NGF-producing cells can be beneficial, it requires surgery with attendant risks of complications and high cost. Intranasal administration (IN) is a non-invasive method that can directly deliver neurotrophins to the CNS [59]. Previous studies [59, 60] have proved that intranasal NGF could bypass the BBB and elicit biological effects. However, the IN NGF concentration in the brain interstitium (perineural fluid that bathes the neuronal cell surfaces), the therapeutic effectiveness, adverse effects and toxicity need further study. So, in our study, we investigated the therapeutic time window for neuroprotective effects of NGF when administered intracerebrally through the perifocal region after focal ischemic injury in rabbits.
Apoptosis contributes to the development of neuronal ischemic infarction after both global [61] and focal [22, 62] ischemia. With more and more researches and understanding in morphology of apoptosis and its biochemical characteristics, we realize that apoptosis is an important form of cell death. Caspase family proteins are known to play a central role in the regulation of apoptosis. Caspase-3, a critical member of the caspase family, accelerates apoptosis and has been suggested as an apoptotic marker [63–65]. Bcl-2 is an integral membrane protein located mainly on the outer membrane of mitochondria. Accumulating evidence indicates that Bcl-2 provides protection against apoptosis by regulating the mitochondrial membrane potential and thereby blocking cytochrome c release into the cytosol [66, 67], where cytochrome c plays a key role in the initiation of apoptosis by activating caspase-3 [68]. Thus, Bcl-2 acts to inhibit cytochrome c translocation, thereby blocking caspase-3 activation and the apoptotic process.
The present study demonstrated that NGF inhibited the expression of pro-apoptotic proteins (caspase-3) and induced that of the anti-apoptotic protein (Bcl-2) in rabbit stroke model, thereby providing the molecular evidence for its neuroprotective activity. In addition, the single NGF administration showed clear neuroprotective and anti-apoptotic activity in rabbit stroke model. These results suggest that NGF showed its therapeutic effect on stroke via directly modulating cellular apoptotic process at the protein expression level.
In summary, NGF is highly efficient in reducing infarct volume and improving neurobehavioral outcome in transient MCAO within the first 5 h. The neuroprotective effect of NGF is time dependent with the best results occurring with administration of NGF as soon after the ischemic injury as possible. Although more studies need to be undertaken to better understand the effects evoked by NGF during neuroprotection, it can be concluded that the inhibition of activation of caspase-3 by increasing the level of anti-apoptotic Bcl-2 is one of the mechanisms underlying the anti-apoptotic effect of NGF on neurons.
References
Chopp M, Zhang ZG, Jiang Q (2007) Neurogenesis, angiogenesis, and MRI indices of functional recovery from stroke. Stroke 38(2 Suppl):827–831
Moonis M, Fisher M (2003) Antiplatelet treatment for secondary prevention of acute ischemic stroke and transient ischemic attacks: mechanisms, choices and possible emerging patterns of use. Expert Rev Cardiovasc Ther 1:611–615
Fisher M, for the Stroke Therapy Academic Industry Roundtable (2003) Recommendations for advancing development of acute stroke therapies: Stroke Therapy Academic Industry Roundtable. Stroke 34:1539–1546
Williams AJ, Berti R, Dave J et al (2004) Delayed treatment of ischemia/reperfusion brain injury extended therapeutic window with the proteosome inhibitor MLN519. Stroke 35:1186–1191
Stroke Therapy Academic Industry Roundtable (STAIR) (1999) Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30: 2752–2758
Levi-Montalcini R (1987) The nerve growth factor 35 years later. Science 237:1154–1162
Mattson MP, Lovell MA, Furukawa K et al (1995) Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem 65:1740–1751
Shimohama S, Ogawa N, Tamura Y et al (1993) Protective effect of nerve growth factor against glutamate-induced neurotoxicity in cultured cortical neurons. Brain Res 632:296–302
Batistatou A, Greene LA (1991) Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol 115:461–471
Inagaki N, Thoenen H, Lindholm D (1995) TrkA tyrosine residues involved in NGF-induced neurite outgrowth of PC12 cells. Eur J Neurosci 7:1125–1133
Segal RA, Greenberg ME (1996) Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci 19:463–489
Cheng B, Mattson MP (1991) NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron 7:1031–1041
Semkova I, Schilling M, Henrich-Noack P et al (1996) Clenbuterol protects mouse cerebral cortex and rat hippocampus from ischemic damage and attenuates glutamate neurotoxicity in cultured hippocampal neurons by induction of NGF. Brain Res 717:44–54
Zhang Y, Tatsuno T, Carney JM et al (1993) Basic FGF, NGF, and IGFs protect hippocampal and cortical neurons against iron-induced degeneration. J Cereb Blood Flow Metab 13:378–388
Shigeno T, Mima T, Takakura K et al (1991) Amelioration of delayed neuronal death in the hippocampus by nerve growth factor. J Neurosci 11:2914–2919
Pechan PA, Yoshida T, Panahian N et al (1995) Genetically modified fibroblasts producing NGF protect hippocampal neurons after ischemia in the rat. Neuroreport 6:669–672
Semkova I, Schilling M, Henrich-Noack P et al (1996) Clenbuterol protects mouse cerebral cortex and rat hippocampus from ischemic damage and attenuates glutamate neurotoxicity in cultured hippocampal neurons by induction of NGF. Brain Res 717:44–54
Tabakman R, Jiang H, Shahar I et al (2005) Neuroprotection by NGF in the PC12 in vitro OGD model. Ann NY Acad Sci 1053:84–96
Shigeno T, Mima T, Takakura K et al (1991) Amelioration of delayed neuronal death in the hippocampus by nerve growth factor. J Neurosci 11:2914–2919
Pechan PA, Yoshida T, Panahian N et al (1995) Genetically modified fibroblasts producing NGF protect hippocampal neurons after ischemia in the rat. Neuroreport 6:669–672
Guégan C, Onténiente B, Makiura Y et al (1998) Reduction of cortical infarction and impairment of apoptosis in NGF-transgenic mice subjected to permanent focal ischemia. Brain Res Mol Brain Res 55:133–140
Yang JP, Liu XF, Liu HJ et al (2008) Extracellular signal-regulated kinase involved in NGF/VEGF-induced neuroprotective effect. Neurosci Lett 434:212–217
Yang JP, Liu HJ, Liu RC (2009) A modified rabbit model of stroke: evaluation using clinical MRI scanner. Neurol Res 31:1092–1096
Manabat C, Han BH, Wendland M et al (2003) Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34:207–213
Purdy PD, Devous MD Sr, Batijer HH et al (1989) Microfibrillar collagen model of canine cerebral infarction. Stroke 20:1361–1367
Schäbitz WR, Hoffmann TT, Heiland S et al (2001) Delayed neuroprotective effect of insulin-like growth factor-I after experimental transient focal cerebral ischemia monitored with MRI. Stroke 32:1226–1233
Linnik MD, Miller JA, Sprinkle-Cavallo J et al (1995) Apoptosis DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Mol Brain Res 32:116–124
Butcher K, Emery D (2010) Acute stroke imaging. Part II: the ischemic penumbra. Can J Neurol Sci 37:17–27
Schaller B, Graf R (2004) Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab 24:351–371
Butte MJ, Hwang PK, Mobley WC et al (1998) Crystal structure of neurotrophin-3 homodimer shows distinct regions are used to bind its receptors. Biochemistry 37:16846–16852
Ibáñez CF (1994) Structure–function relationships in the neurotrophin family. J Neurobiol 25:1349–1361
Robinson RC, Radziejewski C, Spraggon G et al (1999) The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci 8:2589–2597
Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736
Schinder AF, Poo M (2000) The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 23:639–645
Thoenen H (2000) Neurotrophins and activity-dependent plasticity. Prog Brain Res 128:183–191
Tabakman R, Lecht S, Sephanova S et al (2004) Interactions between the cells of the immune and nervous system: neurotrophins as neuroprotection mediators in CNS injury. Prog Brain Res 146:387–401
Barbacid M (1994) The Trk family of neurotrophin receptors. J Neurobiol 25:1386–1403
Kaplan DR, Miller FD (1997) Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol 9:213–221
Gall CM, Isackson PJ (1989) Limbic seizures increase neuronal production of messenger RNA for nerve growth factor. Science 245:758–761
Zafra F, Castrén E, Thoenen H (1991) Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA 88:10037–10041
Batchelor PE, Armstrong DM, Blaker SN et al (1989) Nerve growth factor receptor and choline acetyltransferase colocalization in neurons within the rat forebrain. J Comp Neurol 284:187–204
Hagg T, Manthorpe M, Vahlsing HL et al (1988) Delayed treatment with nerve growth factor reverses the apparent loss of cholinergic neurons after acute brain damage. Exp Neuro 101:303–312
Hagg T, Hagg F, Vahlsing HL et al (1989) Nerve growth factor effects on cholinergic neurons of neostriatum and nucleus accumbens in the adult rat. Neuroscience 30:95–103
Hefti F, Hartikka J, Salvatierra A et al (1986) Localization of nerve growth factor receptors in cholinergic neurons of the human basal forebrain. Neurosci Lett 69:37–41
Montero CN, Hefti F (1988) Rescue of lesioned septal cholinergic neurons by nerve growth factor: Specificity and requirement for chronic treatment. J Neurosci 8:2986–2999
Vantini G, Schiavo N, Martino AD et al (1989) Evidence for a physiological role of nerve growth factor in the central nervous system of neonatal rats. Neuron 3:267–273
Tanaka K, Tsukahara T, Kaku Y et al (1994) Effect of nerve growth factor on delayed neuronal death and microtubule-associated protein 2 after transient cerebral ischaemia in the rat. J Clin Neurosci 1:125–130
Dumont DJ, Fong GH, Puri MC et al (1995) Vascularization of the mouse embryo: a study of flk-1, tek, tie, and vascular endothelial growth factor expression during development. Dev Dyn 203:80–92
Li DQ, Bao YM, Zhao JJ et al (2004) Neuroprotective properties of catalpol in transient global cerebral ischemia in gerbils: dose-response, therapeutic time-window and long-term efficacy. Brain Res 1029:179–185
Wasserman JK, Schlichter LC (2007) Neuron death and inflammation in a rat model of intracerebral hemorrhage: effects of delayed minocycline treatment. Brain Res 1136:208–218
Aronowski J, Strong R, Grotta JC (1997) Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab 17:1048–1056
Muller TB, Haraldseth O, Jones RA et al (1995) Combined perfusion and diffusion-weighted magnetic resonance imaging in a rat model of reversible middle cerebral artery occlusion. Stroke 26:451–457
Maier CM, Sun GH, Kunis D et al (2001) Delayed induction and long-term effects of mild hypothermia in a focal model of transient cerebral ischemia: neurological outcome and infarct size. J Neurosurg 94:90–96
Karibe H, Zarow GJ, Graham SH et al (1994) Mild intraischemic hypothermia reduces postischemic hyperperfusion, delayed postischemic hypoperfusion, blood–brain barrier disruption, brain edema, and neuronal damage volume after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 14:620–627
Jean WC, Spellman SR, Nussbaum ES et al (1998) Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery 43:1382–1396
Saragovi HU, Gehring K (2000) Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol Sci 21:93–98
Thorne RG, Frey WH II (2001) Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet 40:907–946
Krüttgen A, Schneider I, Weis J (2006) The dark side of the NGF family: neurotrophins in neoplasias. Brain Pathol 16:304–310
Frey WH II, Liu J, Chen X et al (1997) Delivery of 125I-NGF to the brain via the olfactory route. Drug Deliv 4:87–92
Zhao HM, Liu XF, Mao XW et al (2004) Intranasal delivery of nerve growth factor to protect the central nervous system against acute cerebral infarction. Chin Med Sci J 19:257–261
Nitatori T, Sato N, Waguri S et al (1995) Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci 15:1001–1011
Li Y, Sharov VG, Jiang N et al (1995) Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol 146:1045–1051
Chen J, Nagayama T, Jin K et al (1998) Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci 18:4914–4928
Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399:A7–A14
Schulz JB, Weller M, Moskowitz MA (1999) Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol 45:421–429
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487
Yang J, Liu X, Bhalla K et al (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Lan X, Qu H, Yao W et al (2008) Granulocyte-colony stimulating factor inhibits neuronal apoptosis in a rat model of diabetic cerebral ischemia. Tohoku J Exp Med 216:117–126
Acknowledgments
This study was supported by the China Ministry of Health Science Foundation (#200310, Dr. Liu), Hebei Natural Science Foundation (#C2010000557, Dr. Yang), Hebei Key Program of Medical Research (#20090101, Dr. Yang) and China Postdoctor Science Foundation (#20070411051, Dr. Yang).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, JP., Liu, HJ., Yang, H. et al. Therapeutic time window for the neuroprotective effects of NGF when administered after focal cerebral ischemia. Neurol Sci 32, 433–441 (2011). https://doi.org/10.1007/s10072-011-0512-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-011-0512-9