
Abstract
Primary adrenal failure comprises an insufficient production of mineralocorticoids and glucocorticoids in the adrenal cortex. A rare manifestation of antiphospholipid syndrome (APS) is adrenal failure. The majority of patients with adrenal involvement in APS develop an irreversible cortisol deficiency and atrophy of the adrenal glands. Adrenal incidentalomas are adrenal masses larger than 1 cm that are discovered in the course of diagnostic evaluation or treatment for another medical condition. Its prevalence is calculated in 1.5–9% of individuals. We describe an exceptional case of a 23-year-old male patient with APS with persistent high levels of antiphospholipid antibodies (aPL) from the time of diagnosis, who developed Addison’s disease as a manifestation of APS with atrophy of the adrenal glands, in whom an adrenal incidentaloma was developed later and was corroborated as an aldosterone-producing adenoma. Currently, the patient is asymptomatic and without manifestations of tumor recurrence. The protumoral effect of elevated and persistent aPL is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The antiphospholipid syndrome (APS) is an autoimmune acquired thrombophilia characterized by the occurrence of recurrent thrombosis and pregnancy morbidity in the presence of antiphospholipid antibodies (aPL) [1]. Primary adrenal failure is the result of the insufficient production of mineralocorticoids and glucocorticoids in the adrenal cortex. Its prevalence ranges from 35 to 60 per million individuals [2]. A rare manifestation of APS is Addison’s disease (AD), which can be the initial presentation of APS or may appear later in the course of the disease. This rare complication has been described in 0.4% of patients with APS [3] and in 10–26% of patients with catastrophic APS [4]. Until 2003, 86 patients with APS and AD had been described [5]; since then, about 30 additional AD-APS cases have appeared. The most recent case was published in 2018 [6]. Thrombosis and hemorrhagic infarction may be caused by local blood stasis, in that the suprarenal gland has multiple arterial supplies, but a single vein renders it more susceptible to venous infarction [7]. Later, an irreversible cortisol deficiency and atrophy of the adrenal glands develop [5, 8]. Adrenal incidentalomas are defined as adrenal masses larger than 1 cm that are discovered in the course of diagnostic evaluation or treatment for another medical condition. Their prevalence is about 1.5–9% and may be found in up to 5% of patients undergoing computed tomography of the abdomen [9]. To date, three cases of incidentalomas in patients with APS have been described [10,11,12]. We present the first case of a patient with primary APS (PAPS) who developed AD with atrophy of the adrenal glands and who subsequently presented an adrenal incidentaloma, with a final diagnosis of an aldosterone-producing adrenal tumor.
Case presentation
A 23-year-old male with an unremarkable history prior to 1995 was admitted to our hospital due to thromboembolism. In 1996, the patient presented deep venous thrombosis (DVT) and pulmonary embolism and was treated with oral anticoagulation without total adherence. Then he presented two more events of DVT, in 1997 and in 1999, at which time a diagnosis of PAPS was established based on the patient’s past clinical history, elevated titers of anticardiolipin antibodies (aCL), and the presence of lupus anticoagulant (LA) corroborated 12 weeks later. After this, the patient resented three thrombotic recurrences in spite of a vena cava filter and oral anticoagulation. In 2008, he was hospitalized due to arterial hypotension (80/60), and later he presented skin hyperpigmentation in face, hands, and oral mucosa, as well as a gradual weight loss of 16 kg. An adrenal CT scan was reported as normal. Blood chemistry revealed the following: serum cortisol 0.7 ng/dL (5–25 ng/dL), TSH 10.46 IU/mL (0.40–4 IU/mL), T3 1.77 ng/dL (84–172 ng/dL), and T4 4.26 ng/dL (0.8–1.8 ng/dL), confirming AD and subclinical hypothyroidism. The patient was started on intravenous (IV) Hydrocortisone followed by oral Prednisone 5 mg daily and Levothyroxine with improvement. In 2010 and 2015, an abdominal CT revealed adrenal atrophy.
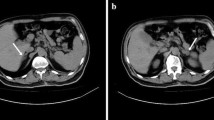
The patient remained asymptomatic with maintenance treatment with Coumadin 4 mg daily (mean international normalized ratio [INR] of 3), Prednisone 5 mg, and Levothyroxine 150 μg/day. He continued showing high titers of aPL (IgG aCL, 117.7 U/mL), with similar values every 2 years and the presence of LA). In 2017, a routine abdominal CT exhibited a right suprarenal tumor that did not demonstrate typical adenoma behavior in contrasted phase and washout (Fig. 1). Hormonal profile (August 2017) showed ACTH 573 pg/mL (chemoluminescence, < 46 pg/mL), cortisol 15.2 μg/dL (chemoluminescence, morning values 5–25 μg/dL), aldosterone 444.1 ng/dL (13.3–231.4 pg/mL), DHEA-S 15 μg/dL (age-adjusted value 136–447.6 μg/dL), and renin 5.8 ng/mL/h (1.5–5.7 ng/mL/h). A suppression test with cortisol revealed a rise in aldosterone levels and a decline in cortisol levels. In April 2018, a laparoscopic adrenalectomy was performed, finding a right adrenal gland of 3 × 2 × 3 cm, of soft consistency and brown discoloration. The histopathological report showed cells rich in lipids that resembled the fascicular zone accompanied by the remains of the adrenal gland without alterations, corroborating the diagnosis of adrenal adenoma (Fig. 2). Hormonal profile after surgery showed an aldosterone level of 22.1 ng/dL. Last hormonal profile in June 2019 showed an aldosterone level of 22.1 ng/dL, testosterone 378 ng/dL (245–1600 ng/dL), cortisol 5.00 μg/dL (5.0–25.0 μg/dL), IgM aCL 21.40 U/mL, and IgG aCL 117.70 U/mL, and prolonged partial thromboplastin time (115 s), without correction after the addition of pool normal plasma, suggesting the persistence of LA, as well as of IgG antiβ2-GP1 antibodies 160 U/mL and IgM 66 U/mL antibodies. Currently, the patient is asymptomatic and without manifestations of tumor recurrence.

Abdominal CT black arrow points the right adrenal adenoma

a Tumor is arranged in nodules of different sizes, monotonous, reminiscent of the fascicular area of the adrenal gland. (HE × 5). b Cells appear concatenated, irregular, with abundant ocher, non-refringent granular pigment (HE × 40)
The search was performed from October to December 2019, based on the searches on PubMed/Medline, Ovid, Science direct, Springer Link, Wiley, and Scopus. The Medical Subject Headings Specific (MeSH) to this research was antiphospholipid syndrome, Addison’s disease, adrenal cortex neoplasms, adrenal insufficiency, and adrenal incidentaloma. These terms were used in logical combinations and phrases (Table 1).
Discussion
The present case represents a classic clinical presentation of thrombotic APS in which the patient developed AD, an infrequent manifestation of APS [6, 8]. The presence of an adrenal incidentaloma in an atrophic gland is rarer. The majority of these tumors are benign and non-functioning, with a minority being functional or malignant [9]. Incidentalomas have been previously described in patients with APS, including one adrenal hematoma, possibly secondary to over-anticoagulation, another with retroperitoneal leiomyosarcoma separated from the adrenal gland, and a case of resistant hypertension with left adrenal mass incidentally discovered and stenosis of multiple arteries due to APS [10,11,12]. In the first two cases, the titers of aPL were not mentioned, and in the latter, positive high titers of anti-β2- glycoprotein I antibodies were reported, but all of the cases corresponded to APS. Clinical manifestations of AD appear when at least 90% of the adrenal cortex has been destroyed and this is generally irreversible, leading to bilateral atrophic glands [8]. The majority of patients with primary hyperaldosteronism have either bilateral idiopathic hyperplasia or unilateral aldosterone-producing adenoma, as in our case [13]. After a diagnosis of primary hyperaldosteronism is established, an adrenalectomy should be performed on patients with a unilateral tumor amenable to surgery [14]. Interestingly, the patient has persistent high levels and is triple-positive for aPL since diagnosis, conferring a high-risk profile [15]. The potential for aPL as an angiogenic factor has been recognized. aPL can induce the expression of vascular endothelial growth factor (VEGF) and tissue factor (TF) in monocytes and endothelial cells [16, 17]. Tissue factor is essential in the coagulation cascade and can enhance angiogenesis through a procoagulant function and platelet activation or via direct activation of protease-activated receptor-2 (PAR2) and proangiogenic signaling in endothelial cells [17]. The protumoral effect of aPL may be related to several mechanisms, including effects on both the tumor and the host cells. Beta 2 glycoprotein I (β2GPI) interacts with multiple regulators of the coagulation/fibrinolysis and angiogenesis pathways and may confer pro- and antiangiogenic effects. On the one hand, it inhibits angiogenesis by interfering with VEGF pathways, and on the other, antibodies against β2GPI might block this process. Angiostatin (AS4.5) is a plasminogen fragment with antitumor and antiangiogenic properties [18]. It is of interest that nicked β2GPI does bind AS4.5 and also inhibits its antiangiogenic effect, thereby promoting angiogenesis [18,19,20]. In this regard, Wu et al. [21] found that aPL accelerated tumor progression in a spontaneous breast cancer model, stimulating the expression of multiple proangiogenic factors in tumor cells from early stages, even in the presence of vascular deficiency. As a result, aPL may promote the transition of indolent tumors to an angiogenic malignant state by means of a TF-mediated pathogenic mechanism. Therefore, the elevation and persistence of aPL could be a novel factor that promotes an angiogenic switch, which may be the possible explanation for the development of an adenoma in a previous atrophic suprarenal gland, as in our patient.
Conclusions
This remarkable case occurred in a patient with adrenal failure secondary to APS with persistent high levels of aPL who developed an aldosterone-producing adenoma, suggesting the protumoral effects of aPL. These mechanisms should be more thoroughly investigated.
References
Cervera R (2017) Antiphospholipid syndrome. Thromb Res 151(Suppl 1):S43–S47. https://doi.org/10.1016/S0049-3848(17)30066-X
Sahin M, Oguz A, Tuzun D, Boysan SN, Mese B, Sahin H, Gul K (2015) Primary adrenal failure due to antiphospholipid syndrome. Case Rep Endocrinol 2015:161497. https://doi.org/10.1155/2015/161497
Cervera R, Boffa MC, Khamashta MA, Hughes GR (2009) The Euro-Phospholipid Project: epidemiology of the antiphospholipid syndrome in Europe. Lupus 18:889–893. https://doi.org/10.1177/0961203309106832
Espinosa G, Cervera R, Font J, Asherson RA (2003) Adrenal involvement in the antiphospholipid syndrome. Lupus 12:569–572. https://doi.org/10.1191/0961203303lu404oa
Espinosa G, Santos E, Cervera R, Piette JC, de la Red G, Gil V, Font J, Couch R, Ingelmo M, Asherson RA (2003) Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients. Medicine (Baltimore) 82:106–118. https://doi.org/10.1097/00005792-200303000-00005
Oliveira D, Ventura M, Melo M, Paiva S, Carrilho F (2018, 2018) Addison’s disease in antiphospholipid syndrome: a rare complication. Endocrinol Diabetes Metab Case Rep. https://doi.org/10.1530/EDM-18-0118
Uthman I, Salti I, Khamashta M (2006) Endocrinologic manifestations of the antiphospholipid syndrome. Lupus 15:485–489. https://doi.org/10.1191/0961203306lu2318rr
Behera KK, Kapoor N, Seshadri MS, Rajaratnam S (2013) Acute adrenal insufficiency due to primary antiphospholipid antibody syndrome. Indian J Endocrinol Metab 17(Suppl 1):S240–S242. https://doi.org/10.4103/2230-8210.119584
Pinto A, Barletta JA (2015) Adrenal tumors in adults. Surg Pathol Clin 8:725–749. https://doi.org/10.1016/j.path.2015.07.005
Khan IN, Adlan MA, Stechman MJ, Premawardhana LD (2015) A retroperitoneal leiomyosarcoma presenting as an adrenal incidentaloma in a subject on warfarin. Case Rep Endocrinol 2015:830814. https://doi.org/10.1155/2015/830814
Kannan S, Satra A, Hamrahian A (2013) Incidental lipid poor adrenal mass in a patient with antiphospholipid syndrome. Case Rep Endocrinol 2013:379852. https://doi.org/10.1155/2013/379852
Marinelli C, Petramala L, Iannucci G et al (2015) Resistant arterial hypertension in a patient with adrenal incidentaloma multiple steno-obstructive vascular lesions and antiphospholipid syndrome. Ann Clin Exp Hypertens 3:1020
Mackay I, Aspinall S (2016) Advances in our understanding of the prognosis of adrenal incidentaloma. Expert Rev Endocrinol Metab 11:529–541. https://doi.org/10.1080/17446651.2016.1233055
Funder JW, Carey RM, Fardella C, Gómez-Sánchez CE, Mantero F, Stowasser M, Young WF Jr, Montori VM, Endocrine Society (2008) Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 93:3266–3281. https://doi.org/10.1210/jc.2008-0104
Tektonidou MG, Andreoli L, Limper M, Amoura Z, Cervera R, Costedoat-Chalumeau N, Cuadrado MJ, Dörner T, Ferrer-Oliveras R, Hambly K, Khamashta MA, King J, Marchiori F, Meroni PL, Mosca M, Pengo V, Raio L, Ruiz-Irastorza G, Shoenfeld Y, Stojanovich L, Svenungsson E, Wahl D, Tincani A, Ward MM (2019) EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis 78:1296–1304. https://doi.org/10.1136/annrheumdis-2019-215213
Cuadrado MJ, Buendía P, Velasco F, Aguirre MA, Barbarroja N, Torres LA, Khamashta M, López-Pedrera C (2006) Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. J Thromb Haemost 4:2461–2469. https://doi.org/10.1111/j.1538-7836.2006.02193.x
Motoki Y, Nojima J, Yanagihara M, Tsuneoka H, Matsui T, Yamamoto M, Ichihara K (2012) Anti-phospholipid antibodies contribute to arteriosclerosis in patients with systemic lupus erythematosus through induction of tissue factor expression and cytokine production from peripheral blood mononuclear cells. Thromb Res 130:667–673. https://doi.org/10.1016/j.thromres.2011.11.048
Nakagawa H, Yasuda S, Matsuura E, Kobayashi K, Ieko M, Kataoka H, Horita T, Atsumi T, Koike T (2009) Nicked β2-glycoprotein I binds angiostatin 4.5 (plasminogen kringle 1-5) and attenuates its antiangiogenic property. Blood 114:2553–2559. https://doi.org/10.1182/blood-2008-12-190629
Yu P, Passam FH, Yu DM, Denyer G, Krilis SA (2008) Beta2-glycoprotein I inhibits vascular endothelial growth factor and basic fibroblast growth factor induced angiogenesis through its amino terminal domain. J Thromb Haemost 6:1215–1223. https://doi.org/10.1111/j.1538-7836.2008.03000.x
Passam FH, Qi JC, Tanaka K, Matthaei KI, Krilis SA (2010) In vivo modulation of angiogenesis by beta 2 glycoprotein I. J Autoimmun 35(3):232–240. https://doi.org/10.1016/j.jaut.2010.06.013
Wu YY, Nguyen A, Wu XX et al (2014) Antiphospholipid antibodies promote tissue factor-dependent angiogenic switch and tumor progression. Am J Pathol 184:3359–3375. https://doi.org/10.1016/j.ajpath.2014.07.027
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Declaration of patient consent
The authors certify that they obtained the patient’s consent for the use of his images and other clinical information reported in the manuscript. The patient understands that his name and initials will not be published, and due efforts will be made to conceal his identity.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Medina, G., Jiménez-Arellano, M.P., Muñoz-Solís, A. et al. Primary antiphospholipid syndrome, Addison disease, and adrenal incidentaloma. Clin Rheumatol 39, 1997–2001 (2020). https://doi.org/10.1007/s10067-020-04978-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-04978-9