
Abstract
Adult-onset Still’s disease (AOSD) is a rare inflammatory disorder of unknown etiology generally characterized by persistent high spiking fever, evanescent rash, and polyarthritis. The pathogenesis of AOSD is only partially known. The pivotal role of macrophage cell activation, which leads to T-helper 1 (Th1) cell cytokine activation, is now well-established in AOSD. Moreover, pro-inflammatory cytokines such as interleukin (IL)-1, -6, and -18 seem to play a key role in this disorder, giving rise to the development of new targeted therapies. For years, treatment of AOSD has been largely empirical, using nonsteroidal anti-inflammatory drugs, corticosteroids, and disease-modifying antirheumatic drugs. Patients with steroid- and methotrexate-refractory AOSD can now benefit from efficient and well-tolerated biologic agents such as IL-1, IL-6, and tumor necrosis factor-α antagonists.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The pathogenesis of adult-onset Still’s disease (AOSD) is not completely understood, but some of the key pro-inflammatory cytokines (interleukin [IL]-1, -18, -6, and tumor necrosis factor [TNF]-α), innate immunity receptors (Toll-like receptors and nucleotide-binding oligomerization-domain-[NOD-]-like receptor family, pyrin domain containing 3 [NLRP3]) and adaptive immunity cells (T-helper 17 [Th17]) are involved. |
IL-1β inhibitors are proven to be effective for AOSD with predominant systemic manifestations but are also effective on articular symptoms. TNF-α blockers and anti-IL-6 agents seem to be more useful for the polyarticular manifestations of the disease. |
1 Introduction
Adult-onset Still’s disease (AOSD) is a rare inflammatory disorder of unknown etiology that is difficult to diagnose because of the heterogeneous clinical presentation [1]. Indeed, the main clinical features (spiking fever, joint involvement, skin rash, and blood neutrophilia), as well as other minor features (pharyngitis, lymph node or spleen enlargement, serositis, myalgia, hepatitis, and abdominal pain), are unspecific [2]. In fact, AOSD is a diagnosis of exclusion, as defined by Yamaguchi classification criteria for the disease [3], and physicians must rule out several likely conditions (autoimmune, infectious, or malignant diseases). AOSD comprises two distinct subgroups with different courses, one with prominent systemic features and one with chronic arthritis (Table 1) [4]. In particular, the systemic form of AOSD is associated with the highest inflammation process, driven by excessive interleukin (IL)-18 and -1β and serum C-reactive protein (CRP) levels, and with the presence of hepatitis and elevated ferritin levels [5]. On the other hand, patients with chronic articular AOSD, with joint involvement principally driven by IL-6 and tumor necrosis factor (TNF)-α, would be at risk of articular destruction [6]. They also present low serum levels of ferritin and IL-18. Patients with systemic AOSD preferentially respond to IL-1β and IL-6 antagonists, whereas those with chronic articular AOSD may also respond to TNF-α blockers [4].
The annual incidence of the disease has been estimated at between 0.16 and 0.4 per 100,000 individuals worldwide, without differences in ethnic groups [7]. Women seem to be slightly more commonly affected than men. Age distribution is bimodal, with one peak between the ages of 15 and 25 years and a second between the ages of 35 and 45 years [8].
AOSD shares common clinical and biological features with another systemic inflammatory condition, systemic juvenile idiopathic arthritis (SJIA), which affects children aged ≤16 years [9]. This supports the hypothesis of a Still’s disease continuum that includes both AOSD and SJIA [10].
Prognosis for AOSD varies widely, ranging from a benign outcome to chronic destructive polyarthritis and/or severe involvement such as pericarditis, myocarditis, hemolytic anemia, acute respiratory distress syndrome, multiple organ failure, thrombotic microangiopathy, central nervous system involvement, and macrophage activation syndrome (MAS; also called reactive hemophagocytic lymphohistiocytosis) [11, 12].
2 Etiopathogenesis
2.1 Genetics
No consistent results have been found in association studies between AOSD and human leukocyte antigen (HLA) loci. This may indicate either a real absence of association or heterogeneity between ethnic groups [6]. In fact, associations with HLA-DR4, HLA-Bw35, or HLA-DRB1 have been occasionally observed in some populations. HLA-Bw35 was the first identified as a susceptibility antigen and associated with a mild pattern of the disease [13]. In Japan and Korea, HLA-DRB1 has been associated with chronic and/or systemic AOSD [6, 14].
Functional polymorphisms in genes encoding innate immunity-associated factors and cytokines, such as IL-18 and macrophage inhibitory factor (MIF), have been described as associated with AOSD [15, 16], but their contribution to the pathogenesis remains vague. In addition, SJIA has been associated with a functional polymorphism in the IL-6 gene promoter and loss-of-function variants in P2RX7, an adenosine triphosphate (ATP) receptor that regulates IL-1 processing and secretion [17, 18].
Recently, a sequencing and quantitative analysis of the genes involved in hereditary autoinflammatory diseases (MEFV, TNFRSF1A, MVK, and NLRP3) in 40 patients with AOSD in Germany observed evidence for a significant association between AOSD and variants in MEFV and TNFRSF1A genes [19].To show a relevant association, systematic studies with bigger cohorts of patients are required.
2.2 Infections
Considering the limited genetic findings, other risk factors will probably be more important as disease-causing or -contributing factors.
The clinical and laboratory features observed in AOSD are highly suggestive of a putative role of infectious agents in the pathogenesis; however, their role as triggers is still under debate. Several viruses and bacteria have been isolated in patients with AOSD [20, 21]. It is proposed that infections can trigger deregulated immune pathways in a genetically predisposed subject. Malignancies can also probably trigger the onset of AOSD, as observed in cases of AOSD-like disease associated with solid cancer and hematological disorders [22].
3 Pathophysiology
Advances in knowledge of the role played by inflammasome in monogenic autoinflammatory diseases have also provided evidence for better understanding of the pathogenic mechanisms of AOSD.
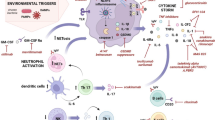
Several external factors such as infectious agents, malignancies, or other environmental factors in patients with genetic predisposition may activate innate immune cells through Toll-like receptors (TLRs), leading first to an abnormal response in innate immunity with cytokine overproduction [23, 24]. Ongoing aberrant IL-1 production would then favor the development of pathogenic adaptive T helper 17 (Th17) cell-mediated responses. Thus, systemic AOSD could evolve from a disease of predominantly autoinflammatory character into a chronic articular one sustained by adaptive immunity (Fig. 1).

Schematic model of adult-onset Still’s disease with a dichotomous view of the disease pathogenesis. Interleukin-1 plays a role in both innate and adaptive immunity, defining evolution from the systemic form to the chronic articular form. AOSD adult-onset Still’s disease, DAMPs danger or damage-associated molecular patterns, IFN interferon, IL interleukin, NK natural killer cell, NLRP3 nucleotide-binding oligomerization-domain-(NOD-) like receptor family, pyrin domain containing 3, PAMPs pathogen-associated molecular patterns, Th17 T-helper 17 cell, TLR7 Toll-like receptor 7
3.1 Innate Immune Cells
Neutrophil and macrophage activation is the hallmark of AOSD. Levels of the neutrophil activation marker cluster of differentiation (CD)-64 (FcγR1) and CXCL-8, a chemokine that mobilizes and activates neutrophils at the site of inflammation, are upregulated in AOSD [25, 26]. Many markers of macrophage activation have correlated well with disease activity. Among those, macrophage-colony stimulating factor (M-CSF) and interferon (IFN)-γ are increased in the serum of patients with AOSD [27, 28]. Calprotectin, macrophage MIF, and intracellular adhesion molecule-1 (ICAM-1) have also been proposed as useful markers for activity and severity in AOSD [29, 30].
The levels and the cytotoxic function of natural killer (NK) cells are reduced in AOSD [31] and improve during efficient treatment of the disease. The impairment of NK cell cytotoxicity could be due to perforin deficiency in the granules of NK and cytotoxic T cells. A decreased perforin expression was in fact reported in SJIA [32].
3.2 Innate Immune System Receptors
TLRs play an important role in the activation of inflammasome, recruitment of neutrophils, and amplification of Th-17-driven inflammatory responses. The TLR7-MyD88 pathway is activated in the dendritic cells of patients with AOSD. Chen et al. [23] showed that elevated levels of TLR7 expression co-existed with elevated transcript and protein levels of MyD88-dependent signaling molecules, including MyD88, IRAK4, and TRAF6, suggesting an activation of TLR7 signaling pathway on peripheral blood mononuclear cells (PBMCs) from patients with AOSD stimulated with TLR7 ligand (imiquimod) [23]. The expression levels of TLR7 positively correlate with disease activity and with serum levels of IL-1β, -6, -18, and IFN-γ. TLR7 levels decrease after effective treatment.
In addition, NLRP3 (nucleotide-binding oligomerization-domain-[NOD-]-like receptor family, pyrin domain containing 3) inflammasome could play a key role in AOSD. Inflammasomes are multiprotein complexes activated by several stimuli as pathogen-associated molecular patterns (PAMPs) or danger- or damage-associated molecular patterns (DAMPs) and subsequent processed IL-1β and IL-18 [33]. No mutation in the NLRP3 gene has yet been associated with AOSD, but recent publications have suggested that an abnormal inflammasome pathway activation could be involved in AOSD pathophysiology [34,35,36]. Even though the precise activators of NLRP3 inflammasome remain unknown in AOSD, recent papers have reported imiquimod, an effective ligand for TLR7, and B19-nonstructural protein (NS)-1, a multifunctional protein of human parvovirus, to be potential activators of the inflammasome [34, 35]. Hsieh et al. [34] also observed significantly higher NLRP3 messenger RNA (mRNA) levels in patients with AOSD, whereas Chen et al. [35] marked a significantly reduced downstream NLRP3 signaling in PBMCs from patients with AOSD treated with NLRP3 inhibitors. Antoniou et al. [36] also provided evidence for an increased NLRP3-mediated IL-1β production in one case of atypical AOSD.
3.3 Adaptive Immunity Cells
CD4+ T-cell activation and proliferation in AOSD is supported by the observed increased concentrations of the α-soluble receptor of IL-2 (CD25) [26]. Patients with AOSD present Th1 subset predominance, which subsequently results in the activation of macrophages and NK cells and promotion of cell-mediated immunity [37]. Furthermore, recent advances have shown higher levels of circulating Th17 cells in patients with active disease and a good correlation between Th17 cells, ferritin levels, and remission after treatment [38].
In addition, the regulatory T-cells (Treg) pathway is an anti-inflammatory mechanism that might be deficient in AOSD. Indeed, circulating CD4+ CD25high Treg and transforming growth factor (TGF)-β are inversely correlated with AOSD activity [37].
3.4 Cytokines/Chemokines
Serum levels of the main pro-inflammatory cytokines are increased in patients with active AOSD, and—of course—levels decrease after treatment with biologic agents targeting specific cytokines [39]. This highlights the crucial role of cytokines and chemokines in the pathogenesis of the disease, even though the cytokine profile is not disease specific.
IL-1β, activated from caspase-1 cleavage via inflammasome, has been implicated in the pathogenesis of AOSD [40]. It is mainly secreted by macrophages, and the serum levels correlate with disease activity. The pivotal role of IL-1β is confirmed by the considerable efficacy of anti-IL-1 therapy in AOSD [4].
IL-18 triggers Th1 response and induces the secretion of IFN-γ in patients with AOSD. High levels of IL-18 are observed in the serum, synovial biopsies, lymph nodes, and liver of patients with AOSD [6, 41]. IL-18 levels have shown good correlation with disease activity, hepatitis, serum ferritin levels, and corticosteroid dependence [42, 43]. IL-18 levels are also significantly higher in patients with AOSD with MAS complication [44]. Recently, Gabay et al. [45] evaluated IL-18 inhibition using recombinant human IL-18 binding protein as a therapeutic option in AOSD (see Sect. 4.4).
Other cytokines, such as IL-6 or TNF-α, that are downstream of IL-1β and IL-18, are also involved in AOSD pathogenesis, possibly as secondary phenomena to the overproduction of IL-1β. IL-6 levels are elevated in patients with AOSD and correlate with skin rash, pyrexia, CRP, ferritin, elevated liver enzymes, and leukocytosis [28, 46]. Interestingly, salmon-colored skin rash specimens revealed heightened IL-6 levels [39].
TNF-α is responsible for synoviocyte proliferation, osteoclastosis, and cachexia. Increased production of TNF-α has been described in AOSD, but the serum and tissue levels did not correlate with disease activity or patient phenotype [28, 39]. However, serum levels of soluble TNF-receptor-2 (sTNF-R2) correlated with serum CRP levels and can potentially be used as a disease activity marker [6].
IL-17 stimulates the production of neutrophil-recruiting chemokines and plays an important role in the development of AOSD-related arthritis. Serum IL-17 derived by Th17 cells was high in patients with AOSD and correlated with Th17-circulating cells [47]. Since both Th17 cells and IL-17 levels decrease upon therapy, Th17-targeted therapies can assume a potential therapeutic role in the management of the disease.
IFN-γ activates macrophages and produces inflammatory cytokines, such as TNF or IL-6. Serum levels of IFN-γ are higher in AOSD with MAS complication [48]. The role of IFN-γ needs to be further defined as animal models seem to indicate that either too much or not enough IFN-γ activity can favor the dichotomy between the systemic and arthritis-prominent forms of AOSD [49, 50].
Moreover, many chemokines are involved in the inflammatory reaction. IL-8/CXCL8 and CX3CL1 are of particular interest in AOSD. IL-8/CXCL8 supports neutrophil recruitment, and its level raises in chronic articular AOSD independent of activity status [26].
CX3CL1 correlates with disease activity and with levels of the serum CRP, ferritin, IL-18, and CD25. It is also able to predict the onset of MAS [51].
4 Therapeutic Strategies
In view of the infrequency of AOSD, most information on treatment management is based on empirical observations through single case reports and small case series. Controlled clinical trials comparing the efficacies of various agents or the usefulness of different therapeutic strategies are lacking. To identify the most appropriate individualized therapeutic strategy for AOSD, as suggested by Govoni et al. [52], it is necessary to consider the disease phase (onset, maintenance, flares), the ongoing predominant clinical features (systemic or articular), and the presence or absence of complications. AOSD therapy aims to control physical signs and symptoms of inflammation, prevent organ failure, and minimize risk of adverse effects. First-line therapy consists of nonsteroidal anti-inflammatory drugs (NSAIDs), especially in the absence of systemic manifestations [53]. However, NSAIDs fail to control the symptoms of AOSD in approximately 80% of patients. Nevertheless, NSAIDs should be considered a supportive treatment during the diagnostic process. High-dose indomethacin (150–250 mg/day), if tolerated, is probably the most effective [54].
Corticosteroids are required in approximately 80% of patients, usually at an initial dose of 0.5–1 mg/kg/day [55]. Response to corticosteroids occurs within a couple of hours or a few days. However, patients treated with a high dose of prednisone (0.8 mg/kg/day) achieve faster remissions and have fewer relapses than those who receive lower doses [56].
Unfortunately, about 45% of patients develop steroid dependence with mild or severe side effects. Thus, disease-modifying antirheumatic drugs (DMARDs) and immunosuppressive agents are often required, especially if therapy fails. Methotrexate or cyclosporine A have a steroid-sparing effect in both systemic and articular clinical forms [53, 57]. However, the available evidence supporting the use of DMARDs in AOSD comes from small case series or case reports.
Biologic therapy with different mechanisms of action have been increasingly and successfully used in the last 15 years to treat AOSD. Available evidence consists of single case reports and case series, also relating to a considerable cohort of patients [58]. The rationale to use biologics, actually targeting cytokines, lies in increased knowledge about the pathophysiology of the disease. It is further possible, although not yet proven, that early treatment with biologic agents may take advantage of a “window of opportunity” in which disease pathophysiology can be altered to avoid the chronic evolution of AOSD. Finally, biologic therapy should be considered a first step in severe or life-threatening systemic manifestations.
4.1 TNF-α Inhibitors
TNF-α inhibitors, such as infliximab, etanercept, and adalimumab, were the first biologic agents used in AOSD in single case reports or small series of patients in the early 2000 s [59]. Overall, TNF-α inhibitors are well-tolerated, even if their efficacy seems to be limited in time.
Cavagna et al. [60] were the first to describe the use of infliximab in three patients with AOSD showing a rapid and prolonged efficacy of both articular and systemic symptoms and also a marked steroid-sparing effect.
Etanercept is a recombinant soluble form of the human 75-kd TNF-receptor fusion protein. In a small case series of 12 AOSD cases refractory to DMARDs, etanercept 25 mg biweekly for 6 months led to arthritis improvement in half of the patients with no adverse events [61].
These positive results with TNF-α inhibitors were not confirmed by Fautrel et al. [62], who observed complete remission in only five patients in a French observational study of 20 cases with refractory AOSD treated with infliximab and etanercept.
Data on adalimumab, a fully humanized monoclonal antibody targeted against TNF-α, are limited. It was used successfully in AOSD that did not respond to etanercept but failed to improve the clinical outcomes in two other cases [63, 64].
Switching from one TNF-α inhibitor to another may be useful, as demonstrated in case of partial response or secondary loss of response [65], although infliximab appears to be more effective than etanercept. Moreover, Maria et al. [4] observed a longer retention rate of TNF-α inhibitors in chronic polyarticular refractory AOSD than in the systemic form of the disease.
4.2 IL-1 Inhibitors
IL-1 is a key cytokine in the immunopathogenesis of AOSD and thus an important target in the therapeutic approach. Nowadays, three IL-1 inhibitors are in use: anakinra, canakinumab, and rilonacept.
Anakinra (IL-1 receptor antagonist [IL-1Ra]) is a recombinant, nonglycosylated form of human IL-1Ra that acts as a pure receptor antagonist binding to the IL-1 receptor (IL-1RI) and preventing activation of this receptor by either IL-1β or IL-1α [66]. It has a short half-life (4–6 h), so is administered subcutaneously daily, generally at 100 mg/day. A lower dose is prescribed in clinical remission, generally 50 mg/day or 100 mg every 2–3 days [67]. A growing number of reports describe a rapid response to anakinra characterized by impressive reduction in disease activity, fever resolution, and normalization of hematologic parameters within a few days after the first injection [68,69,70]. However, relapses occurred within a few days of treatment discontinuation.
Several studies in SJIA suggest that anakinra is more effective when administered early in the course of the disease [71]. Laskari et al. [72] observed that 84% of patients with AOSD treated early with anakinra achieved a rapid and sustained remission after a year of follow-up. Overall, despite the lack of randomized controlled trials in AOSD, anakinra seems to have a rapid and sustained efficacy, especially if used in combination with steroids [73]. Anakinra seems to be more effective in patients with highly active systemic AOSD than in those with isolated chronic arthritis [74]. In case of insufficient response due to its short half-life, anakinra can be increased to 200 mg split into twice-daily administrations.
Canakinumab is a fully human monoclonal antibody against IL-1β with a half-life of 26 days, which makes it possible to administer canakinumab 150 mg every 4–8 weeks. Canakinumab has a well-established safety and efficacy profile in SJIA, as reported by recently published SJIA clinical trials (G2305 and G2301) [75]. Treatment with canakinumab resulted in rapid and sustained improvement of both articular and systemic features [76]. In 2016, the drug was approved by the European Medicines Agency (EMA) and the US FDA for a license extension to treat both SJIA and AOSD, supported by the concept of a Still’s disease continuum that includes both juvenile and adult-onset forms [11]. Recently, Feist et al. [77] described, in a subgroup of patients with SJIA aged ≥16 years representative of AOSD patients, efficacy, safety, and exposure-response relationships of canakinumab similar to those in children and adolescents with SJIA. A multicenter, placebo-controlled, 12-week trial (NCT02204293) of the efficacy, safety, and tolerability of canakinumab in 68 patients with AOSD will provide more robust efficacy data. Unfortunately, the results from the trial of canakinumab in adult patients were not available at the time of publication.
Rilonacept (IL-1 trap molecule) is a construct of two extracellular chains of the IL-1R complex (IL-1R plus IL-1RAP) fused to the Fc portion of human immunoglobulin G1 (IgG1). Since it contains both receptor components, rilonacept is able to bind IL-1β and IL-1α with high affinity. Its half-life is longer than that of anakinra, and it is administered at 160 mg/week followed by regular or on-demand maintenance doses. It is used in AOSD refractory to NSAIDs and DMARDs and sometimes also to anakinra [78, 79]. It has shown impressive efficacy, with rapid and sustained control of systemic and articular symptoms.
4.3 IL-6 Antagonists
IL-6 serum levels are commonly increased in active AOSD. Thus, IL-6 should be a suitable target for the treatment of refractory cases. Tocilizumab is a humanized anti-IL-6 receptor antibody that recognizes both membrane-bound and soluble forms of IL-6 receptor, specifically blocking IL-6. It is administered subcutaneously at doses of up to 8 mg/kg every 2 weeks [80].
Several case reports have highlighted the potential efficacy of tocilizumab in refractory AOSD [81,82,83]. Moreover, a randomized placebo-controlled trial (NCT00642460) demonstrated the efficacy of tocilizumab in 85% of patients with SJIA [84]. Impressive improvements were attained in both clinical and laboratory parameters [85]. Beneficial effects are better documented with the chronic pattern of the disease, but tocilizumab is also effective on systemic symptoms [80]. As described for infliximab and anakinra, tocilizumab yielded a marked steroid-sparing effect. It has been reported and reviewed that tocilizumab has been effective for AOSD complicated with MAS and systemic inflammatory response syndrome [86, 87], even though MAS should also be recognized as a complication of tocilizumab treatment [88].
4.4 Other Treatments
Intravenous immunoglobulins (IVIGs), a pool of immunoglobulin G (IgG), has been administered to patients with AOSD at the usual dose of 2 g/kg in 2–5 days every month. In two open-label studies, IVIGs were shown to be effective and well-tolerated in half of the patients [89, 90]. However, a controlled study in SJIA failed to demonstrate efficacy [91].
Abatacept is a costimulation modulator that inhibits T-lymphocyte activation by binding to CD80 and CD86 and blocking interaction with CD28. Two patients with disease refractory to other biologics were successfully treated with abatacept [92, 93], but it failed in two other patients [83, 84].
Rituximab is a chimeric monoclonal anti-CD20 antibody that induces depletion of peripheral B cells. A few case reports have shown a positive response in refractory AOSD to rituximab 375 mg/m2 given twice at 4-week intervals [94, 95]. However, rituximab failed to treat some other patients with resistant AOSD [96].
Tadekinig alfa is the drug name for recombinant human IL-18-binding protein (IL-18BP). A recent open-label, dose-finding French trial (NCT02398435) tested this agent in 23 patients with active AOSD. Two dose cohorts (80 and 160 mg) were treated for 12 weeks and followed-up for 4 more weeks. The study results suggested that the 80 mg dose had a meaningful clinical efficacy in “difficult to treat” disease as early as week 3 [45]. The authors also demonstrated that tadekinig alfa had a favorable safety and tolerability profile, with only mild adverse events at the injection site.
5 Conclusion
Recent insights into the pathophysiology of AOSD categorized it as a complex autoinflammatory disorder. The pathogenesis of AOSD remains incompletely understood, but some key pro-inflammatory cytokines (IL-1, IL-18, IL-6, and TNF-α), innate immunity receptors (TLRs and NLPR3), and adaptive immunity cells (Th17) are involved. Advances in the understanding of the immunopathogenesis of AOSD has led to the use of new biologic agents, which have proved effective in the management of refractory AOSD and make it possible to distinguish between systemic and chronic articular disease patterns.
Traditional DMARDs have limited efficacy in controlling systemic manifestations of AOSD. On the other hand, IL-1 inhibitors are effective as steroid-sparing drugs and in inducing and maintaining disease remission. Furthermore, anakinra and canakinumab are the only drugs approved by the FDA and EMA for AOSD.
Bearing in mind the increased knowledge about the pathophysiology of AOSD, there is a clear rationale to use biologics targeting IL-1. Nonetheless, further investigation is needed to determine whether these drugs should be considered as first-step agents and whether such a treatment in a “window of opportunity” may prevent the chronic evolution of AOSD.
References
Kadavath S, Efthimiou P. Adult-onset Still’s disease-pathogenesis, clinical manifestations, and new treatment options. Ann Med. 2015;47:6–14.
Sfriso P, Priori R, Valesini G, Rossi S, Montecucco CM, D’Ascanio A, et al. Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016;35:1683–9.
Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424–30.
Maria AT, Le Quellec A, Jorgensen C, Touitou I, Rivière S, Guilpain P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: dichotomous view for cytokine and clinical expressions. Autoimmun Rev. 2014;13:1149–59.
Pouchot J, Fautrel B. Maladie de Still de l’adulte. In: Piette JC, Meyer O, Sibilia J, editors. Médecine/Sciences F. Maladies et syndromes systémiques, 5ème édition Paris: Médecine Sciences Flammarion; 2008; vol. 1. p. 449–68.
Fujii T, Nojima T, Yasuoka H, Satoh S, Nakamura K, Kuwana M, et al. Cytokine and immunogenetic profiles in Japanese patients with adult Still’s disease. Assoc Chron Articul Dis Rheumatol (Oxford). 2001;40:1398–404.
Magadur-Joly G, Billaud E, Barrier JH, Pennec YL, Masson C, Renou P, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54:587–90.
Wakai K, Ohta A, Tamakoshi A, Ohno Y, Kawamura T, Aoki R, et al. Estimated prevalence and incidence of adult Still’s disease: findings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7:221–5.
Evensen KJ, Nossent HC. Epidemiology and outcome of adult-onset Still’s disease in Northern Norway. Scand J Rheumatol. 2006;35:48–51.
Jamilloux Y, Gerfaud-Valentin M, Martinon F, Belot A, Henry T, Seve P. Pathogenesis of adult-onset Still’s disease: new insights from the juvenile counterpart. Immunol Res. 2015;61:53–62.
Nirmala N, Brachat A, Feist E, Blank N, Specker C, Witt M, et al. Gene-expression analysis of adult-onset Still’s disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr Rheumatol Online J. 2015;13:50.
Sampalis JS, Esdaile JM, Medsger TA, Partridge AJ, Yeadon C, Senécal JL, et al. A controlled study of the long-term prognosis of adult Still’s disease. Am J Med. 1995;98:384–8.
Terkeltaub R, Esdaile J, Décary F, Harth M, Lister J, Lapointe N. HLA-Bw35 and prognosis in adult Still’s disease. Arthritis Rheum. 1981;12:1469–72.
Joung CI, Lee HS, Lee SW, Kim CG, Song YH, Jun JB, et al. Association between HLA-DR B1 and clinical features of adult onset Still’s disease in Korea. Clin Exp Rheumatol. 2003;21:489–92.
Sugiura T, Kawaguchi Y, Harigai M, Terajima-Ichida H, Kitamura Y, Furuya T, et al. Association between adult-onset Still’s disease and interleukin-18 gene polymorphisms. Genes Immun. 2002;3:394–9.
Wang FF, Huang XF, Shen N, Leng L, Bucala R, Chen SL, et al. A genetic role for macrophage migration inhibitory factor (MIF) in adult-onset Still’s disease. Arthritis Res Ther. 2013;15:R65.
Fishman D, Faulds G, Jeffery R, Mohamed-Ali V, Yudkin JS, Humphries S, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. 1998;102:1369–76.
Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–15.
Sighart R, Rech J, Hueber A, Blank N, Löhr S, Reis A, et al. Evidence for genetic overlap between adult onset Still’s disease and hereditary periodic fever syndromes. Rheumatol Int. 2018;38:111–20.
Wouters JM, van der Veen J, van de Putte LB, de Rooij DJ. Adult onset Still’s disease and viral infections. Ann Rheum Dis. 1988;47:764–7.
Balleari E, Cutolo M, Accardo S. Adult-onset Still’s disease associated to toxoplasma gondii infection. Clin Rheumatol. 1991;10:326–7.
Liozon E, Ly KH, Vidal-Cathala E, Fauchais AL. Adult-onset Still’s disease as a manifestation of malignancy: report of a patient with melanoma and literature review. Rev Med Interne. 2014;35:60–4.
Chen DY, Lin CC, Chen YM, Lan JL, Hung WT, Chen HH, et al. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthritis Res Ther. 2013;15:R39.
Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13:708–22.
Komiya A, Matsui T, Nogi S, Iwata K, Futami H, Takaoka H, et al. Neutrophil CD64 is upregulated in patients with active adult-onset Still’s disease. Scand J Rheumatol. 2012;41:156–8.
Choi JH, Suh CH, Lee YM, Suh YJ, Lee SK, Kim SS, et al. Serum cytokine profiles in patients with adult onset Still’s disease. J Rheumatol. 2003;30:2422–7.
Matsui K, Tsuchida T, Hiroishi K, Tominaga K, Hayashi N, Hada T, et al. High serum level of macrophage-colony stimulating factor (M-CSF) in adult-onset Still’s disease. Rheumatology (Oxford). 1999;38:477–8.
Hoshino T, Ohta A, Yang D, Kawamoto M, Kikuchi M, Inoue Y, et al. Elevated serum interleukin 6, interferon-gamma, and tumor necrosis factor-alpha levels in patients with adult Still’s disease. J Rheumatol. 1998;25:396–8.
Zou YQ, Lu LJ, Li SJ, Zeng T, Wang XD, Bao CD, et al. The levels of macrophage migration inhibitory factor as an indicator of disease activity and severity in adult-onset Still’s disease. Clin Biochem. 2008;41:519–24.
Chen DY, Lan JL, Lin FJ, Hsieh TY. Association of intercellular adhesion molecule-1 with clinical manifestations and interleukin-18 in patients with active, untreated adult-onset Still’s disease. Arthritis Rheum. 2005;53:320–7.
Lee SJ, Cho YN, Kim TJ, Park SC, Park DJ, Jin HM, et al. Natural killer T cell deficiency in active adult-onset Still’s Disease: correlation of deficiency of natural killer T cells with dysfunction of natural killer cells. Arthritis Rheum. 2012;64:2868–77.
Wulffraat NM, Rijkers GT, Elst E, Brooimans R, Kuis W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology (Oxford). 2003;42:375–9.
Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–35.
Hsieh CW, Chen YM, Lin CC, Tang KT, Chen HH, Hung WT, et al. Elevated expression of the NLRP3 inflammasome and its correlation with disease activity in adult-onset still disease. J Rheumatol. 2017;44:1142–50.
Chen DY, Chen YM, Chen HH, Hsieh CW, Gung NR, Hung WT, et al. Human parvovirus B19 nonstructural protein NS1 activates NLRP3 inflammasome signaling in adult-onset Still’s disease. Mol Med Rep. 2018;17:3364–71.
Antoniou KM, Margaritopoulos GA, Giannarakis I, Choulaki C, Fountoulakis N, Siafakas NM, et al. Adult onset Still’s disease: a case report with a rare clinical manifestation and pathophysiological correlations. Case Rep Med. 2013;2013:981232.
Chen DY, Lan JL, Lin FJ, Hsieh TY, Wen MC. Predominance of Th1 cytokine in peripheral blood and pathological tissues of patients with active untreated adult onset Still’s disease. Ann Rheum Dis. 2004;63:1300–6.
Chen DY, Chen YM, Chen HH, Hsieh CW, Lin CC, Lan JL. The associations of circulating CD4+CD25 high regulatory T cells and TGF- b with disease activity and clinical course in patients with adult-onset Still’s disease. Connect Tissue Res. 2010;51:370–7.
Chen DY, Lan JL, Lin FJ, Hsieh TY. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J Rheumatol. 2004;31:2189–98.
Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–90.
Rooney T, Murphy E, Benito M, Roux-Lombard P, FitzGerald O, Dayer J-M, et al. Synovial tissue interleukin-18 expression and the response to treatment in patients with inflammatory arthritis. Ann Rheum Dis. 2004;63:1393–8.
Priori R, Barone F, Alessandri C, Colafrancesco S, McInnes IB, Pitzalis C, et al. Markedly increased IL-18 liver expression in adult-onset Still’s disease-related hepatitis. Rheumatology (Oxford). 2011;50:776–80.
Kawashima M, Yamamura M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001;44:550–60.
Maruyama J, Inokuma S. Cytokine profiles of macrophage activation syndrome associated with rheumatic diseases. J Rheumatol. 2010;37:967–73.
Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77:840–7.
Scheinberg MA, Chapira E, Fernandes ML, Hubscher O. Interleukin 6: a possible marker of disease activity in adult onset Still’s disease. Clin Exp Rheumatol. 1996;14:653–5.
Chen DY, Chen YM, Lan JL, Lin CC, Chen HH, Hsieh CW. Potential role of Th17 cells in the pathogenesis of adult-onset Still’s disease. Rheumatology (Oxford). 2010;49:2305–12.
Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105:1648–51.
Canna SW. Interferon-γ: friend or foe in systemic juvenile idiopathic arthritis and adult Still’s Disease. Arthritis Rheumatol. 2014;66:1072–6.
Avau A, Mitera T, Put S, Put K, Brisse E, Filtjens J, et al. Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with Freund’s complete adjuvant: regulation by interferon-γ. Arthritis Rheumatol. 2014;66:1340–51.
Kasama T, Furuya H, Yanai R, Ohtsuka K, Takahashi R, Yajima N, et al. Correlation of serum CX3CL1 level with disease activity in adult-onset Still’s disease and significant involvement in hemophagocytic syndrome. Clin Rheumatol. 2012;31:853–60.
Govoni M, Bortoluzzi A, Rossi D, Modena V. How I treat patients with adult onset Still’s disease in clinical practice. Autoimmun Rev. 2017;16:1016–23.
Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62:2530–5.
Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. QJM. 1986;61:1055–65.
Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, et al. Adult-onset Still disease: manifestations, treatments, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93:91–9.
Kim YJ, Koo BS, Kim YG, Lee CK, Yoo B. Clinical features and prognosis in 82 patients with adult-onset Still’s disease. Clin Exp Rheumatol. 2014;32:28–33.
Pouchot J, Sampalis JS, Beaudet F, Carette S, Decary F, Salusinsky-Sternbach M, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70:118–36.
Colafrancesco S, Priori R, Valesini G, Argolini L, Baldissera E, Bartoloni E, et al. Response to interleukin-1 inhibitors in 140 italian patients with adult-onset still’s disease: a multicentre retrospective observational study. Front Pharmacol. 2017;8:369.
Al-Homood IA. Biologic treatments for adult-onset Still’s disease. Rheumatology (Oxford). 2014;53:32–8.
Cavagna L, Caporali R, Epis O, Bobbio-Pallavicini F, Montecucco C. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin Exp Rheumatol. 2001;19:329–32.
Husni ME, Maier AL, Mease PJ, Overman SS, Fraser P, Gravallese EM, et al. Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum. 2002;46:1171–6.
Fautrel B, Sibilia J, Mariette X, Combe B. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64:262–6.
Benucci M, Li GF, Del Rosso A, Manfredi M. Adalimumab (anti-TNF-alpha) therapy to improve the clinical course of adult-onset Still’s disease: the first case report. Clin Exp Rheumatol. 2005;23:733.
Rech J, Ronneberger M, Englbrecht M, Finzel S, Katzenbeisser J, Manger K, et al. Successful treatment of adult-onset Still’s disease refractory to TNF and IL-1 blockade by IL-6 receptor blockade. Ann Rheum Dis. 2011;70:390–2.
Aikawa NE, Ribeiro AC, Saad CG, Pereira RM, Levy M, Silva CA, et al. Is anti-TNF switching in refractory Still’s disease safe and effective? Clin Rheumatol. 2011;30:1129–34.
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–32.
Ortiz-Sanjan F, Blanco R, Riancho-Zarrabeitia L, Castañeda S, Olivé A, Riveros A, et al. Efficacy of anakinra in refractory adult-onset Still’s disease: multicenter study of 41 patients and literature review. Medicine (Baltimore). 2015;94:e1554.
Kone-Paut I, Piram M. Targeting interleukin-1 b in CAPS (cryopyrin-associated periodic) syndromes: what did we learn? Autoimmun Rev 2012;12:77e80.
Giampietro C, Fautrel B. Anti-interleukin-1 agents in adult onset still’s disease. Int J Inflam. 2012;2012:317820.
Lequerré T, Quartier P, Rosellini D, Alaoui F, De Bandt M, Mejjad O, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67:302–8.
Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–55.
Laskari K, Tzioufas AG, Moutsopoulos HM. Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res Ther. 2011;13:R91.
Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis SN. Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann Rheum Dis. 2007;66:842–3.
Guignard S, Dien G, Dougados M. Severe systemic inflammatory response syndrome in a patient with adult onset Still’s disease treated with the anti-IL1 drug anakinra: a case report. Clin Exp Rheumatol. 2007;25:758–9.
Ruperto N, Quartier P, Wulffraat N, Woo P, Ravelli A, Mouy R, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557–67.
Kontzias A, Efthimiou P. The use of Canakinumab, a novel IL-1 b long-acting inhibitor, in refractory adult-onset Still’s disease. Semin Arthritis Rheum. 2012;42:201–5.
Feist E, Quartier P, Fautrel B, Schneider R, Sfriso P, Efthimiou P, et al. Efficacy and safety of canakinumab in patients with Still’s disease: exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. Clin Exp Rheumatol. 2018. (Epub ahead of print).
Petryna O, Cush JJ, Efthimiou P. IL-1 trap rilonacept in refractory adult onset Still’s disease. Ann Rheum Dis. 2012;71:2056–7.
Henderson C, Wilson M, Pham TH, Dolan G, Gobbo A, Snyder C, et al. Safety and efficacy of IL-1 trap in resistant adult onset Still’s disease: 24 month follow-up of open label treatment and biomarkers of response. Arthritis Rheum. 2010;62:1831.
Ortiz-Sanjuan F, Blanco R, Calvo-Rio V, Narvaez J, Rubio Romero E, Olive A, et al. Efficacy of tocilizumab in conventional treatment-refractory adult-onset Still’s disease: multicenter retrospective open-label study of thirty-four patients. Arthritis Rheumatol. 2014;66(6):1659–65.
Cipriani P, Ruscitti P, Carubbi F, Pantano I, Liakouli V, Berardicurti O, et al. Tocilizumab for the treatment of adult-onset Still’s disease: results from a case series. Clin Rheumatol. 2014;33:49–55.
Elkayam O, Jiries N, Dranitzki Z, Kivity S, Lidar M, Levy O, et al. Tocilizumab in adult-onset Still’s disease: the Israeli experience. J Rheumatol. 2014;41:244–7.
Puéchal X, DeBandt M, Berthelot JM, Breban M, Dubost JJ, Fain O, et al. Tocilizumab in refractory adult Still’s disease. Arthritis Care Res. 2011;63:155–9.
De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95.
Ma Y, Wu M, Zhang X, Xia Q, Yang J, Xu S, et al. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: a meta-analysis. Mod Rheumatol. 2018;8:1–9.
Watanabe E, Sugawara H, Yamashita T, Ishii A, Oda A, Terai C. Successful tocilizumab therapy for macrophage activation syndrome associated with adult-onset Still’s disease: a case-based review. Case Rep Med. 2016;2016:5656320.
Masui-Ito A, Okamoto R, Ikejiri K, Fujimoto M, Tanimura M, Nakamori S, et al. Tocilizumab for uncontrollable systemic inflammatory response syndrome complicating adult-onset Still disease: case report and review of literature. Medicine (Baltimore). 2017;96:e7596.
Tsuchida Y, Sumitomo S, Shoda H, Kubo K, Fujio K, Yamamoto K. Macrophage activation syndrome associated with tocilizumab treatment in adult-onset Still’s disease. Mod Rheumatol. 2017;27:556–7.
Permal S, Wechsler B, Cabane J, Perrot S, Blum L, Imbert JC. Treatment of Still disease in adults with intravenous immunoglobulins. Rev Med Interne. 1995;16:250–4.
Vignes S, Wechsler B, Amoura Z, Papo T, Francès C, Huong DL, et al. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol. 1998;16:295–8.
Prieur AM. Intravenous immunoglobulins in Still’s disease: still controversial, still unproven. J Rheumatol. 1996;23:797–800.
Ostrowski RA, Tehrani R, Kadanoff R. Refractory adult-onset still disease successfully treated with abatacept. J Clin Rheumatol. 2011;17:315–7.
Quartuccio L, Maset M, De Vita S. Efficacy of abatacept in a refractory case of adult-onset Still’s disease. Clin Exp Rheumatol. 2010;28:265–7.
Ahmadi-Simab K, Lamprecht P, Jankowiak C, Gross WL. Successful treatment of refractory adult onset Still’s disease with rituximab. Ann Rheum Dis. 2006;65:1117–8.
Bartoloni E, Alunno A, Luccioli F, Santoboni G, Gerli R. Successful treatment of refractory adult-onset Still’s disease with anti-CD20 monoclonal antibody. Clin Exp Rheumatol. 2009;27:888–9.
Giampietro C, Ridene M, Lequerre T, Costedoat Chalumeau N, Amoura Z, Sellam J, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res (Hoboken). 2013;65:822–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
Paolo Sfriso, Sara Bindoli, and Paola Galozzi have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Sfriso, P., Bindoli, S. & Galozzi, P. Adult-Onset Still’s Disease: Molecular Pathophysiology and Therapeutic Advances. Drugs 78, 1187–1195 (2018). https://doi.org/10.1007/s40265-018-0956-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0956-9