
Abstract
SCA48 is a novel spinocerebellar ataxia (SCA) originally and recently characterized by prominent cerebellar cognitive-affective syndrome (CCAS) and late-onset ataxia caused by mutations on the STUB1 gene. Here, we report the first SCA48 case from Turkey with novel clinical features and diffusion tensor imaging (DTI) findings, used for the first time to evaluate a SCA48 patient. A 65-year-old female patient with slowly progressive cerebellar ataxia, cognitive impairment, behavioral changes, and a vertical family history was evaluated. Following the exclusion of repeat expansion ataxias, whole exome sequencing (WES) was performed. Brain magnetic resonance imaging (MRI), including DTI, and single-photon emission computed tomography (SPECT) were used to study the primarily affected tracts and regions. WES revealed the previously reported heterozygous truncating mutation in ubiquitin ligase domain of STUB1 (ENST00000219548:c.823_824delCT, ENSP00000219548:p.L275Dfs*16) leading to a frameshift. Patient’s cognitive status was compatible with CCAS. Novel clinical features different from the original report include later onset chorea, dystonia, general slowness of movements, apraxia, and palilalia, some of which have been recently reported in two families with different STUB1 mutations. CCAS is a prominent and often early feature of SCA48 which may be followed years after the onset of the disease by other complex neurological signs and symptoms. DTI may be helpful for demonstrating the cerebello-frontal tracts, involved in CCAS-associated SCA48, the differential diagnosis of which may be challenging especially in its early years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxias (SCAs) are a group of neurodegenerative disorders with dominant inheritance patterns. To date, almost 50 different types of SCAs have been described, the most recent one being SCA48 reported by Genis et al. in a Spanish family [1]. In this family with a dominant inheritance pattern, patients present initially with cognitive and behavioral symptoms compatible with cerebellar cognitive-affective syndrome (CCAS), later followed by cerebellar motor signs. A more recent paper by De Michele et al. described in two Italian families, two heterozygous STUB1 mutations resulting in an expanded phenotype which starts with ataxia and later includes complex extrapyramidal features [2].
Here, we report a SCA48 family from Turkey, the third study worldwide and the second with the original Spanish mutation. The initial clinical feature in our family is CCAS, which is followed years later by motor cerebellar symptoms but other than the Spanish family additionally by extrapyramidal signs, further expanding the phenotype of the disease. In addition to conventional MRI, we applied the MRI-based neuroimaging technique, diffusion tensor imaging (DTI), which makes it possible to estimate the white matter organization of the brain [3]. Our study reveals for the first time the tracts involved in CCAS-associated SCA48.
Case report
Clinical findings
A 65-year-old female patient was taken to the neurology outpatient clinic by her family with complaints of gait imbalance, decreased verbal output, dysarthric speech with redundancy, forgetfulness, and behavioral changes. She had no systemic complaints.
At the age of 51, she was initially evaluated at another center because of complaints of diminished speech fluency, deficits in attention and judgement, inappropriate answers, and mild amnesia with slow progression and anxiety. Her repeated neuropsychiatric evaluations, at the age of 53 and 55, indicated progressive frontal lobe dysexecutive syndrome with prominent attention deficits. At that time, her cranial MRI revealed mild cerebral and slightly more prominent cerebellar atrophy (data not shown).
The cognitive and behavioral symptoms of the patient were followed 3 years later by impaired fine motor skills and 8 years later by imbalance of gait. Her speech became slurred, and she began to repeat the same words and sentences for several times. Throughout the last 4 years, she also developed severe anxiety. One year prior to her admittance at the age of 64, she developed aggressive behavior, delusions, frequent awakenings during night sleep, mild involuntary movements on her lower extremities, and urinary incontinence. Loss of appetite and weight in the last 6 months along with inadequate self-care and hygiene were reported. The speed of the progression of the disease accelerated in the last 5 years.
Her examination during admittance showed prominent cognitive decline with language impairment and amnesia. She was unable to perform the neuropsychological tests, to read, and to write. She was anosognostic towards her disease being unaware of her condition. She had ideomotor apraxia (video 1), her reaction time to stimuli was also delayed. She had cerebellar dysarthria and a limited verbal output accompanied by palilalia.
Her gait was ataxic. Sometimes, she needed unilateral support for walking. She had a slightly bent posture with postural instability, global slowness of movements, and parkinsonian type of shuffling gait without accompanying rigidity and resting tremor (video 2). She had bilateral pyramidal findings with hyperreflexia and positive Hoffmann and Babinski signs. Mild facial and cervical dystonia were observed (videos 3 and 4). She also had choreic movements on her extremities, more prominent on the feet (video 5).
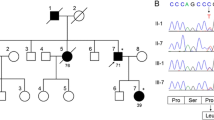
The mother (II-4) and maternal aunt (II-2) of our index case (III-7) were not personally examined by us; however, her daughter, a physician, provided a very detailed description of their disease and disease course (Fig. 1). Their disease also began with cognitive symptoms and much later complemented by cerebellar signs, altogether lasting for over 10 years. The severity of their symptoms also showed an accelerating course with additional neurological features like chorea and dystonia towards the terminal stage of the disease. They both passed away bedridden and cachectic. Further demographic and clinical features of the family are listed in (Table 1).

Pedigree. Index patient (III-7) AO = 51; mother (II-4) AO = 60 AEX = 80; aunt (II-2) AO = 60 AEX = 77 (AO, age of onset; AEX, age of exitus)
Neuroimaging findings
Brain MRI with DTI and 99mTc-HMPAO Brain perfusion SPECT imaging were performed. MRI revealed cerebrocerebellar atrophy, more prominent in the cerebellum (Fig. 2). Cerebellar atrophy was localized especially to segments VI–VII of the posterior vermis. Atrophy of the frontal and occipital lobes was more pronounced compared with other cerebral regions. DTI showed paucity of the cerebellar connections to the brain stem and cerebrum, especially in the superior longitudinal fasciculus. Superior and middle cerebellar peduncles were mildly atrophic. Subcortical U-fibers at the frontal dorsolateral region were not visible bilaterally (Fig. 3). Brain SPECT showed hypoperfused areas in frontal, parietal, and temporal lobes. Cerebellar involvement was more prominent on the right side (Fig. 4).

T2-weighted axial plane and sagittal plane images of the patient (top panel). Atrophy at frontal and occipital lobes (filled arrows) is present. Temporal lobes are spared (a, b). Cerebellar atrophy localized to segments VI–VII is shown (plain arrows) (b, c). T2-weighted axial plane and sagittal plane images of the age- and sex-matched control (bottom panel)

Diffusion tractography images of the index patient (a–d) and healthy individual at the same age (e–h). a, e Middle cerebellar peduncles (green arrows) and cerebellar connections (white arrows) are severely affected. b, f Superior cerebellar peduncles (blue arrows) and cerebellar connections (white arrows) are mildly atrophic compared with healthy individual. c, g Superior longitudinal fascicules (red arrows) are severely affected. c, g Subcortical U-fibers at frontal dorsolateral regions (yellow area) are not visible bilaterally. d, h After the capsular structures in the corticocerebellar tracts were marked with ROI (region of interest) the images were taken. Fiber tractography images demonstrated that frontal long tracts and subcortical U-fiber tracts seemed to be prominantly affected (pink arrow)

99mTc-HMPAO brain perfusion SPECT images of the index patient (a and b) and of a healthy control (c and d). 99mTc-HMPAO brain perfusion SPECT shows the hypoperfusion areas in right parietal lobe supramarginal gyrus, left frontal lobe superior precentral gyrus, right frontal lobe middle and inferior gyrus, left temporal lobe superior and middle gyrus, and right mid-temporal gyrus in cerebrum and decreased perfusion in both cerebellum, more prominent on the right.
Genetic findings
After testing for Huntington’s disease, DRPLA, C9orf72, and SCA17, we performed whole exome sequencing analysis.
Methods
Conventional screening for dominant ataxias, Huntington’s disease and C9orf72
The DNA was isolated from whole blood. PCR amplification followed by fragment analysis and the Sanger sequencing were applied for screening for dominant SCA17 and DRPLA and Huntington’s disease (HD). RP-PCR was conducted for C9orf72 analysis.
Next-generation sequencing, bioinformatic evaluation, and validation by the Sanger analysis
Whole exome sequencing (WES) was performed (Macrogen, Korea) on index patient (III-7).
Raw data obtained from WES was processed using the SEQ platform (Genomize, Istanbul, Turkey) and the identified variant in the STUB1 gene confirmed by the Sanger sequencing.
Results
The sample tested was negative for the most frequent dominant SCAs, C9orf72, and HD. WES revealed an already reported heterozygous CT deletion in exon 7 of STUB1 (c.823_824delCT) leading to a frameshift and to a pre-mature termination after 16 codons [1]. The deletion was confirmed by the Sanger sequencing (Fig. 5).

The Sanger chromatogram of patient sample. Highlighted CT corresponds to the position of the deletion and the start of the frameshift
Discussion
CCAS was first described by Schmahmann and Sherman [4] with impairment in executive function and visual-spatial memory, personality changes characterized by blunting of affect or disinhibited, inappropriate behavior, and difficulties with language production such as mild anomia, agrammatism, and dysprosodia. CCAS is the result of damage to the cognitive cerebellum in the cerebellar posterior lobe (esp lobulus VI, VII). CCAS may be seen isolated or with other cerebellar symptoms [4, 5] and becomes clinically evident after the onset of cerebellar motor symptoms in most hereditary ataxias [6, 7]
The Turkish SCA48 family presented, sharing the same truncating mutation with the first SCA48 family, confirms the original description of the disease with the onset of cognitive affected syndrome (CAS), slow progressive course, and follow-up motor cerebellar signs. Our family expands the clinical spectrum of the original STUB1 mutation causing SCA48 beyond cerebellar features, including late-onset chorea, dystonia, and parkinsonism. Further novel clinical findings in our patient include early-onset progressive ideomotor apraxia and severe palilalia (Table 1).
Although cerebellar atrophy was an early and prominent finding in our index patient’s MRIs, cerebellar ataxia followed years after cognitive and affective symptoms as a late manifestation of the disease. This clinical presentation was challenging in the early years of the disease and thus was interpreted as signs of primary degenerative dementia.
The SPECT findings of our patient displayed pronounced dysfunction in the right cerebellar hemisphere, associated with hypoperfusion of the left fronto-cortical areas. In a right-handed individual, linguistic region in the left cortical hemisphere is linked to the right posterior lateral cerebellar hemisphere, which may also explain the prominent language difficulties of our patient [8]. All symptomatic Genis and De Michele cases showed cerebellar atrophy on MRIs. Moreover, even one asymptomatic case from the Genis family revealed cerebellar atrophy involving the posterior area of the vermis and paravermis (lobes VI and VII).
Diffusion tensor imaging data on ataxias in general is scarce and this is the first application of DTI to SCA48, as a complex ataxia, with involvement of cognitive and accompanying cerebellar findings. The application of DTI to SCA48 in this study, revealing severe disconnection between the cerebellum and frontal lobes, together with bilateral involvement of dentate nuclei, supports CCAS. Our DTI findings indicate that diffusion tractography imaging may be a promising technique in the future in demonstrating the cerebello-frontal tracts, involved not only in CCAS-associated SCA48 (the differential diagnosis of which may be challenging especially in its early years) but also in many other spinocerebellar ataxias [9, 10].
STUB1 gene encodes the protein CHIP, which is one of the main components of the cellular protein quality control system by its co-chaperone and ubiquitin ligase functions [11, 12]. STUB1 is expressed most ubiquitiously in the cerebellum and frontal lobes in the brain (GTEX database, https://www.gtexportal.org/home/gene/STUB1) [13]. Recessive mutations in STUB1 were associated with SCAR16 [14,15,16,17,18,19]. Homozygous deleterious mutations located in the U-box protein domain of STUB1 lead to cognitive impairment [14, 15]. More precisely, a functional analysis revealed that a recessive mutation in this domain induces a loss of ubiquitin ligase activity but does not affect the co-chaperone activity [20]. As opposed to SCA48, in which cognitive impairment is an early and prominent manifestation of the disease, cognitive impairment is either absent [14] or it is an accompanying mild or sometimes moderate late symptom of the disease phenotype [13, 15, 19].
In the study of Genis et al., a heterozygous mutation in the U-box protein domain leads to a pre-mature stop codon (p.L275Dfs*16) and is responsible for cognitive and ataxic neurodegeneration. The same monoallelic deletion is present in our patient, however, with an expanded phenotype. This expanded phenotype is also observed in the De Michele study, but most importantly with a different temporal profile. The widespread degeneration pattern, including the striatum, is remarkable.
STUB1 has been shown to result in multi-system neurodegeneration coinciding with systemic involvement other than central nervous system, such as hypogonadism, short stature, presbyacusis, ulcerative colitis, and type 1 diabetes mellitus in recessive disease [1, 19]. It is interesting to note that dominant STUB1 mutations can give rise to widespread neurodegeneration, however, without systemic involvement. The heterozygously inherited pathogenic mutations in SCA48 have to be further investigated in order to shed light on their effects on disease initiation and mechanisms.
With the definition of SCA48, the importance of non-motor function of the cerebellum is further confirmed. In patients with a dominant family history and adult-onset CCAS without initial motor cerebellar symptoms, but cerebellar atrophy on MRI or affected cerebello-frontal tracts on DTI, SCA48 should be considered as a possible diagnosis. Cerebellar motor symptoms of the disease may develop years after, along with late-onset chorea, dystonia, apraxia, and general slowness of movements as revealed by the case presented.
References
Genis D, Ortega-Cubero S, San Nicolás H, Corral J, Gardenyes J, de Jorge L, López E, Campos B, Lorenzo E, Tonda R, Beltran S, Negre M, Obón M, Beltran B, Fàbregas L, Alemany B, Márquez F, Ramió-Torrentà L, Gich J, Volpini V, Pastor P (2018) Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91:e1988–e1998. https://doi.org/10.1212/WNL.0000000000006550
De Michele G, Lieto M, Galatolo D, Salvatore E, Cocozza S, Barghigiani M, Tessa A, Baldacci J, Pappatà S, Filla A, De Michele G, Santorelli FM (2019) Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2019.05.001
O’Donnell LJ, Westin C-F (2011) An introduction to diffusion tensor image analysis. Neurosurg Clin N Am 22:185–196, viii. https://doi.org/10.1016/j.nec.2010.12.004
Schmahmann JD, Sherman JC (1998) The cerebellar cognitive affective syndrome. Brain J Neurol 121(Pt 4):561–579
Schmahmann JD (2019) The cerebellum and cognition. Neurosci Lett 688:62–75. https://doi.org/10.1016/j.neulet.2018.07.005
Durr A (2010) Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 9:885–894. https://doi.org/10.1016/S1474-4422(10)70183-6
Soong B-W, Morrison PJ (2018) Spinocerebellar ataxias. Handb Clin Neurol 155:143–174. https://doi.org/10.1016/B978-0-444-64189-2.00010-X
Mariën P, Borgatti R (2018) Chapter 11 - Language and the cerebellum. In: Huisman TAGM (ed) Manto M. Elsevier, Handbook of Clinical Neurology, pp 181–202
Li H, Ma J, Zhang X (2014) Diffusion tensor imaging of spinocerebellar ataxia type 12. Med Sci Monit Int Med J Exp Clin Res 20:1783–1791. https://doi.org/10.12659/MSM.891104
Adanyeguh IM, Perlbarg V, Henry P-G, Rinaldi D, Petit E, Valabregue R, Brice A, Durr A, Mochel F (2018) Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. NeuroImage Clin 19:858–867. https://doi.org/10.1016/j.nicl.2018.06.011
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19:4535–4545. https://doi.org/10.1128/mcb.19.6.4535
Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C, Pearl LH (2005) Chaperoned ubiquitylation--crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell 20:525–538. https://doi.org/10.1016/j.molcel.2005.09.023
GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45:580–585. https://doi.org/10.1038/ng.2653
Shi Y, Wang J, Li J-D, Ren H, Guan W, He M, Yan W, Zhou Y, Hu Z, Zhang J, Xiao J, Su Z, Dai M, Wang J, Jiang H, Guo J, Zhou Y, Zhang F, Li N, Du J, Xu Q, Hu Y, Pan Q, Shen L, Wang G, Xia K, Zhang Z, Tang B (2013) Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS ONE 8. https://doi.org/10.1371/journal.pone.0081884
Synofzik M, Schüle R, Schulze M, Gburek-Augustat J, Schweizer R, Schirmacher A, Krägeloh-Mann I, Gonzalez M, Young P, Züchner S, Schöls L, Bauer P (2014) Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts. Orphanet J Rare Dis 9:57. https://doi.org/10.1186/1750-1172-9-57
Heimdal K, Sanchez-Guixé M, Aukrust I, Bollerslev J, Bruland O, Jablonski GE, Erichsen AK, Gude E, Koht JA, Erdal S, Fiskerstrand T, Haukanes BI, Boman H, Bjørkhaug L, Tallaksen CM, Knappskog PM, Johansson S (2014) STUB1 mutations in autosomal recessive ataxias – evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis 9:1–12. https://doi.org/10.1186/s13023-014-0146-0
Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA (2014) Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology 83:287–288. https://doi.org/10.1212/WNL.0000000000000600
Depondt C, Donatello S, Simonis N, Rai M, van Heurck R, Abramowicz M, D’Hooghe M, Pandolfo M (2014) Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology 82:1749–1750. https://doi.org/10.1212/WNL.0000000000000416
Bettencourt C, de Yébenes JG, López-Sendón JL, Shomroni O, Zhang X, Qian S-B, Bakker IMC, Heetveld S, Ros R, Quintáns B, Sobrido M-J, Bevova MR, Jain S, Bugiani M, Heutink P, Rizzu P (2015) Clinical and neuropathological features of spastic ataxia in a Spanish family with novel compound heterozygous mutations in STUB1. Cerebellum Lond Engl 14:378–381. https://doi.org/10.1007/s12311-014-0643-7
Shi C-H, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao C-Y, True C, Wang R-H, Wang Q-Z, Sun S-L, Seminara SB, Patterson C, Xu Y-M (2014) Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet 23:1013–1024. https://doi.org/10.1093/hmg/ddt497
Acknowledgments
We would like to thank our technical assistants Irmak Şahbaz and Müge Koç Kovancılar, and our graduate students for excellent technical assistance in the laboratory and during the writing of the manuscript. We are very grateful to the daughter of our index case, a physician, for her continuous assistance and support in supplying us in an academic fashion on her mother’s, grandmother’s, and grandaunt’s disease stories. Last not least, we would like to wholeheartedly acknowledge Koç University, KUTTAM, and Suna and Inan Kıraç Foundation for creating a great research environment for us and for their generous support of the study.
Ethical approval
All procedures performed were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individuals included in this study. Additional informed consent was obtained from all individual participants from whom identifying information is included in this article.
Author information
Authors and Affiliations
Contributions
R. Palvadeau and Z. E. Kaya-Güleç contributed equally to this work. Başak A. N and Ertan S contributed equally to this work.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Palvadeau, R., Kaya-Güleç, Z.E., Şimşir, G. et al. Cerebellar cognitive-affective syndrome preceding ataxia associated with complex extrapyramidal features in a Turkish SCA48 family. Neurogenetics 21, 51–58 (2020). https://doi.org/10.1007/s10048-019-00595-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-019-00595-0