
Abstract
Background
Spinal cerebellar ataxia 11 (SCA11) is a rare disease, characterized by progressive cerebellar ataxia, abnormal eye sign. Four families have been reported in the past. We report on China’s first family with spinocerebellar ataxia 11.
Methods
A careful investigation of the clinical manifestations, brain imaging, and exome and Sanger sequencing were utilized to identify pathogenic genetic variants in a three-generation pedigree that includes 5 affected individuals.
Results
The proband and affected members began to develop cerebellar ataxia, dysarthria, nystagmus, and strabismus at approximately age 40 for no apparent reason. The lifespan of patients in the family is shortened. Brain MRIs showed cerebellar atrophy and slight atrophy of the bulbar medulla. Electromyography showed extensive neurogenic damage. Sensory evoked potentials of lower limbs showed damage to the spinal-brainstem-cortical conduction pathway. Genetic analysis revealed a novel point mutation (c.3290T>C) in the TTBK2 gene encoding tau-microtubule kinase 2, which led to an amino acid exchange (p.Val1097Ala). The missense mutation segregated with the phenotype. The mutation has a very low mutation rate in the population, the variant amino acids are highly conserved among species, and protein function damage prediction at the mutation site is detrimental and is highly likely to cause protein damage. The pathogenicity prediction of the mutation site shows that it is likely to cause disease. This variation is consistent with the diagnosis of SCA11.
Conclusion
The first SCA11-affected family in China was characterized by gait instability, movement disorders and dysarthria with obvious cerebellar atrophy. The pathogenic allele was a c.3290T>C mutation in the TTBK2 gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Hereditary ataxia is a group of hereditary diseases characterized by the slow development of uncoordinated gait, often accompanied by poor coordination of hands, language, and eye movements. The cerebellum often shrinks. Hereditary ataxia can be autosomal dominant, autosomal recessive, or X-linked [1]. Genetic heterogeneity is prominent and there are multiple known loci and genes linked to the disease. Spinocerebellar ataxias (SCAs) have irregular distributions worldwide. SCA1 is the most common and no SCA3 has been identified in Poland [2]. A recent genetic analysis of hereditary spinocerebellar ataxia (SCA) in mainland China showed that SCA3 is essentially the most common subtype of ADCA with sporadic patients in mainland China. However, no pathogenic mutations were found for the SCA5, SCA11, SCA13, SCA14, SCA27, and SCA31 subtypes [3].
TTBK2 is a multifunctional kinase composed of 1244 amino acids that is involved in important cellular processes. Its main function is to modify specific targets to initiate the formation of cilia [4, 5]. SCA11 is caused by mutations in the tau-tubulin kinase 2 encoding gene TTBK2 [6]. TTBK2 is highly expressed in Purkinje cells, granule, the hippocampus, midbrain, and the substantia nigra in the cerebellum, while its expression is low in the cortex. Affected brain tissues show substantial cerebellar degeneration and tau deposition [6, 7]. TTBK2 phosphorylates tau protein at sites 208 and 210 [8]. Furthermore, TTBK2 may interact with the inositol/IP3 pathway, and stabilize cells (especially Purkinje) to resist calcium-induced cell death [7, 9]. The mechanism of tau toxicity is still unclear, and the interaction between TTBK2 and tau may be related to the common pathway of TTBK2, gsk-3β and tau [6].
Spinal cerebellar ataxia type 11 (SCA11) is exceedingly rare. Its clinical features include slow progression of aggravated purely cerebellar disorders with a normal life expectancy, with symptoms such as ataxia of the gait, ataxia of the trunk, gallop at full speed, upper extremity ataxia, lower extremity ataxia, dysarthria, and horizontal nystagmus [10]. Currently, only 5 affected families have been identified, one each from Britain, Pakistan, France, Germany, and Denmark, and their TTKB2 mutations are not completely the same. These include c.1329_1330insA from Britain, c.1284_1285delAG from Pakistan, c. 1306_1307delGA from Germany and France, and c.1205_1207delinsA from Denmark [6, 10, 11]. These mutations are all code shift mutations, leading to the premature generation of termination codons in the mRNA and ultimately to protein truncation and/or nonsense-mediated decay of the mutant transcript, leading to haploinsufficiency of the TTBK2 protein [7, 11, 12].
Brain MRI scans showed cerebellar atrophy, as well as moderate atrophy of the pons and medulla. Macroscopic examination showed retention of the brainstem with obvious cerebellar atrophy. Microscopically, the cerebellum showed severe loss of Purkinje cells and significant reduction of granule, a large number of empty baskets, and proliferation of Bergmann glial cells [6].
SCA3 is the most common autosomally inherited cerebellar ataxia in China, while SCA11 is very rare [1, 13]. Here, we report the first family with spinocerebellar ataxia 11 in China and a new TTBK2 mutation. In this study, we carefully collected and analyzed the clinical data of the proband and her family members, including clinical manifestations, imaging results, electrophysiological examinations, and next generation sequencing (NGS), which detected a new pathogenic TTBK2 mutation and helped us diagnose the first case of familial SCA11 in China.
Materials and methods
Family pedigree and patient evaluation
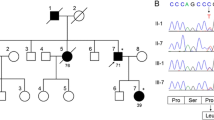
All the examined persons were from the same Chinese SCA11-affected family. At the time of writing, there were 26 persons from 3 generations, including 10 patients with ataxia and/or maldevelopment, as shown in Fig. 1. All investigations were conducted with informed consent as part of the clinical diagnostic assessment.

Pedigree of the first Chinese SCA11-affected family. The arrow indicates the patient; circles indicate female and squares indicate male family members; solid symbols indicate affected family members; + and – signs indicate individuals with and without the TTBK2 mutation, respectively; the / symbols indicate deceased family members
Clinical data collection
After signing the informed consent form, the proband underwent a test for serum creatine kinase, electromyography (EMG; Keypoint electromyography/evoked potential instrument, Denmark), and brain magnetic resonance imaging (MRI) (SingaHDxt 3.0T magnetic resonance scanner; GE, USA). Two family members (III6 and III8) completed the testing for serum creatinine kinase (CK) and electromyography after signing the informed consent forms. III6 also completed the brain magnetic resonance imaging (MRI) examination.
Next-generation sequencing
The whole-exome sequencing of the proband revealed three suspicious variants: c.1541A>G in ATXN7 exon 9, c.580G>A in DARS2 exon 6, and c.3290T>C in TTBK2 exon 15. Then, next-generation sequencing was used to assess the corresponding genes in family members who volunteered to participate in the examination, and it was found that all the members with similar clinical manifestations to those of the proband had a heterozygous TTBK2 c.3290T>C mutation, while no ATXN7 or DARS2 mutations were found.
Results
Clinical description
The proband was a Chinese woman born in 1969. She was transferred to our department in 2018 and diagnosed with hereditary ataxia due to progressive dysarthria and walking instability in the past 5 years. Her father, uncle, aunt, grandmother, brothers, and sisters also had similar ataxia symptoms, so she was suspected of having heritable ataxia. Her mother was healthy and had no neurological symptoms. From the age of 44, the proband gradually developed slurred speech and unsteady walking without an obvious proximal trigger. Physical examination findings include the following: dysarthria, liquid dysphagia, nystagmus, gait indicative of cerebellar ataxia, both open and closed eyes positive for Romberg’s sign, limb muscle strength level 5, muscle atrophy, active tendon reflex in the limbs, negative pathological signs.
Some of her siblings were also showing similar symptoms. The main symptoms were as follows: The younger sister (III6) of the proband had disease onset at the age of 38, presenting with dysarthria, gait indicative of cerebellar ataxia, and weakness of both lower limbs after activity. Physical examination for nystagmus and dysdiadokokinesia was positive. At the age of 37, III15 had already suffered from articulation disorder, cerebellar ataxia gait; physical examination findings include the following: nystagmus, finger nose test (+), right lower limb muscle strength 5-, active tendon reflex of both lower limbs. At the age of 43, III8 developed progressive dysarthria, liquid dysphagia, cerebellar ataxia, gait, limb weakness, and movement disorder. The brother (III9) of the proband developed dysarthria and cerebellar ataxia gait at the age of 42. The father (II2), uncles (II3, II7), aunt (II5), and grandmother (I2) of the proband also developed the disease around the age of 40, with progressive dysarthria, unstable walking, choking and liquid dysphagia, limb weakness, limb muscle atrophy, and finally complete immobility. The uncles, aunts, and grandmothers (II3, II7, II5, I2) all died around the age of 50, whereby II5 and I2 died by suicide. The specific causes of death were unknown, although the affected persons seemed to have shorter than normal life expectancy for the years in question. However, it could not be ruled out whether they also suffered from some other diseases, including mental illness.
Brain imaging
The brain magnetic resonance imaging of the proband showed that the sulcus and cistern were widened and deepened, the cerebellum was moderately atrophied, there was mild atrophy of the medulla oblongata, and no obvious abnormalities in the brain MRA. There was no obvious abnormality in brain magnetic resonance imaging and MRA of III6 (Fig. 2a, b).

The proband’s brain MRI showed the sulcus and cistern were widened and deepened, especially in the cerebellum (a) and the medulla oblongata was slightly atrophied (b)
Laboratory examinations
Laboratory examination showed that the proband had a serum creatine kinase (CK) concentration of 109.52 U/L (normal range 38-174 IU/L); III6 had serum CK of 305.53 U/L, and III8 had 136.37 U/L. We only tested serum creatine kinase (CK) once in these patients, so no trend was observed.
Electrophysiological examination
Electromyography of the proband showed extensive neurogenic damage. Sensory evoked potentials of the lower limbs indicated an impaired spinal cord-brainstem-cortex conduction pathway. However, repeated nerve stimulation (RNS) and the electroencephalogram were normal. In III6, the EMG was indicative of generalized neurogenic damage, while the RNS showed no characteristic changes. In III8, the EMG was indicative of generalized neurogenic damage (lower limbs affected).
Genetic analysis
Genomic DNA was isolated from whole blood samples using a Flexigene DNA kit. Whole-exome sequencing was conducted for the proband. At least 0.5 μg of DNA from each of the subjects was fragmented into 180-280 bp segments using a Covaris sonicator. Qubit 2.0 and Agilent 2100 were used for preliminary quantification and detection of the library insert size. qPCR was used for accurate quantification of the effective concentration to ensure the library quality, and an Illumina HiSeq X Ten was utilized for library sequencing. The Burrows-Wheeler Alignment tool (BWA) was used to match the clean reads without adapters or debased reads to the human reference genome (UCSC hg19). Single nucleotide variation (SNV) and InDel (small insert-deletion variation) were identified by GATK (v3.6), and CODEX, XHMM (v1.0), and KSCNV were used to analyze the possible copy number. The variants were filtered with SNP database (dbSNP), 1000 Genomes Project, the Exome Aggregation Consortium (Exac), and Exome Sequencing Project (ESP) 6500. Sorting Intolerant from Tolerant (SIFT) and Polymorphism Phenotyping version2 (PolyPhen-2) were employed to predict protein function. PhyloP, phastCons, and GERP++ are used for conservative analysis. Annotate variation (ANNOVAR) software was used to annotate the locations of variation in genes and transcripts.
Targeted next-generation sequencing identified a characteristic heterozygous variation in the nucleotide number 3290 of the TTBK2 gene coding region from T to C (c.3290T>C), which resulted in the amino acid, No.1097 being changed from Val to Ala. The same mutation was present both in the proband (III3) and in the affected family members III6, III8, III9, and III15, while the unaffected member III11 did not have the same mutation. This is a missense mutation, and the mutation and its pathogenicity have not been reported (refer to the PubMed and HGMD Prodatabases). The gene sequencing results of the proband and family members are shown in Fig. 3. Due to conditional constraints, we were unable to detect the mutation rate of this site in the normal population. The new mutation is not a polymorphism change. By searching the public database of human genetic variation (reference database 1000Genomes and Exac database), we found that the frequency of new mutation sites in the general population and East Asian population is very low. The specific data are shown in Supplementary Table 1. Using ANNOVAR, we found that the mutation site is harmful and may lead to protein function damage. The specific data are shown in Supplemenatry Table 2. The amino acid of TTBK2 gene protein No.1097 is highly conserved among different species. The database data are shown in Table 1.

First-generation sequencing: TTBK2 sequence, an arrow indicating the mutation (c.3290T>C)
Discussion
SCA11 presents with progressive cerebellar ataxia and abnormal eye sign (jerky pursuit, horizontal, vertical nystagmus). Occasionally, a pyramidal tract sign, peripheral neuropathy, and dystonia may be seen. The average age of onset was from the early teens in patients of Pakistani origin to between 40 and 50 years of age in French and German families. The age of onset in the family we diagnosed was about 40 years old, and the clinical manifestations were similar to those of the previously found SCA11-affected families, including ataxia, maldevelopment, nystagmus, and strabismus. The diagnosis of SCA11 mainly depends on clinical manifestations and the presence of mutations in TTBK2, which is the only known gene whose mutations lead to SCA11.
SCA11 patients can show cerebellar atrophy in brain magnetic resonance imaging (MRI). We found atrophy of the cerebellum and medulla in the magnetic resonance imaging of the proband, but no cerebellar atrophy was seen in the image of the sister. It is not clear which factors are related to the severity and development of the disease. We suspect that the differences in imaging results may be related to the age of onset, current age, progress, and severity of each case. Our neurophysiological EMG measurements of three members of the family showed extensive neurogenic damage, so EMG testing may provide some help in the diagnosis of the disease. The previous SCA11-affected families showed that the disease does not affect life expectancy, whereby many patients had a lifespan of more than 75 years. However, many members of the family from this study died at around 50 years old, which seems to be shorter than the normal life expectancy at the time in question in China. It should also be noted that two of them died by suicide. The specific causes of death of the other members are not clear, and we are not sure whether these members may have suffered from other diseases, including mental, cognitive, psychological, and physical diseases. We should strengthen the management of the comorbidities of the disease and conduct neuropsychological tests to detect the comorbidities of patients early and begin psychological counseling and other related treatments where necessary.
This is the first SCA11-affected family found in China, which broadens our knowledge of the phenotypic spectrum of this rare disease. The previously discovered TTKB2 mutations in SCA11-affected families were c.1329_1330insA in the UK, c.1284_1285delAG in Pakistan, c.1306_1307delGA in Germany and France, and c.1205_1207delinsA in Denmark. These mutations are frame-shift mutations [6, 10, 11]. By contrast, the TTBK2 mutation we found here is a new point mutation (c.3290T>C) that leads to an amino acid exchange (p.Val1097Ala). In one study, a patient with ataxia was found to have a heterozygous TTBK2 c.2525A>G (exon 14) mutation that resulted in the amino acid exchange of p.E842G [14]. In the SCA11 screening studies in Germany and France, a German-Polish family was found to have exon 13 point mutation of TTBK2 (c.1952C>T, p.T651M). However, none of these mutations are considered pathogenic due to lack of genetic test results for affected family members or the mutation is not separated from the phenotype [11].
We found a new heterozygous TTBK2 mutation, c.3290T>C (exon 15), which leads to an amino acid exchange (p.Val1097Ala). This mutation was found in the similarly affected III8, III9, III6, and III15, but not in the disease-free III11. This missense mutation segregated with the phenotype. The occurrence frequency of the new mutation in the general population is very low, and the pathogenicity prediction SIFT scores, PolyPhen scores, and MutationTaster scores are high, and the mutated amino acids are highly conserved among different species. We found through the HOPE web service that the wild-type and mutated amino acids have different sizes, and the mutants have smaller residues. The wild-type residue is predicted (using the Reprof software) to be located in its preferred secondary structure, a β-strand. The mutant residue prefers to be in another secondary structure; therefore, the local conformation will be slightly destabilized. Although we did not perform functional tests, we believe that the mutation is likely to be pathogenic. Unfortunately, genetic testing was not performed on all members of the family, especially a sufficient number of clinically negative members. Continued follow-up of the family will help us be more specific about the disease-causing mutations. Functional testing should be done in the following studies, since it will help us further understand and determine the pathogenic mechanism of the mutation and further study the relationship between SCA11 and TTBK2 mutations.
Further identification of TTBK2 mutations, especially truncation and missense mutations, will be of great significance for the understanding of SCA11. In the future, we can obtain knowledge about the type of TTBK2 mutations and the clinical effects of various variants by studying more pedigrees, so as to provide a solid foundation for genetic diagnosis of SCA11.
Abbreviations
- SCA11:
-
Spinocerebellar ataxia 11
- SCA3:
-
Spinocerebellar ataxia 3
- ADCA:
-
Autosomal dominant cerebellar ataxia
- TTBK2:
-
tau-tubulin kinase2
- IP3:
-
inositol triphosphate
- gsk-3β:
-
Glycogen synthase kinase-3β
- MRI:
-
Magnetic resonance imaging
- NGS:
-
next-generation sequencing
- EMG:
-
electromyography
- CK:
-
creatinine kinase
- ATXN7 :
-
The pathogenic gene of spinocerebellar ataxia-7
- DARS2 :
-
Pathogenic genes of leukocephalopathy with brain stem and spinal cord involvement and lactate elevation
- MRA:
-
Magnetic resonance angiography
- EMG:
-
Electromyography
- RNS:
-
repeated nerve stimulation
References
Bird T D . Hereditary ataxia overview [M]// GeneReviews™. PubMed, 2008. DOI:https://doi.org/10.1038/gim.2013.28
Krysa Wioletta, Sulek Anna, Rakowicz Maria et al. High relative frequency of SCA1 in Poland reflecting a potential founder effect.[J] .Neurol Sci, 2016, 37: 1319–1325. DOI:https://doi.org/10.1007/s10072-016-2594-x
Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, Guan W, Pan Q, Xia K, Tang B, Jiang H (2011) Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. [J]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36(6):482–489. https://doi.org/10.3969/j.issn.1672-7347.2011.06.003
Anderson KV (2012) The spinocerebellar ataxia-associated gene tau tubulin kinase 2 controls the initiation of ciliogenesis. [J]. Cell 151(4):847–858. https://doi.org/10.1016/j.cell.2012.10.010
Jung-Chi L, Tony YT, Roc WR et al (2015) TTBK2: a tau protein kinase beyond tau phosphorylation[J]. Biomed Res Int 2015:1–10. https://doi.org/10.1155/2015/575170
Houlden H, Johnson J, Gardner-Thorpe C, Lashley T, Hernandez D, Worth P, Singleton AB, Hilton DA, Holton J, Revesz T, Davis MB, Giunti P, Wood NW (2008) Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. [J]. Nat Genet 39(12):1434–1436. https://doi.org/10.1038/ng.2007.43
Ikezu S, Ikezu T (2014) Tau-tubulin kinase. Front Mol Neurosci 7:33. https://doi.org/10.3389/fnmol.2014.00033
Kitanotakahashi M, Morita H, Kondo S et al (2007) Expression, purification and crystallization of a human tau-tubulin kinase 2 that phosphorylates tau protein[J]. Acta Crystallogr Sect F Struct Biol Cryst Commun 63(7):602–604. https://doi.org/10.1107/S1744309107028783
Leemput JVD, Chandran J, Knight MA et al (2007) Deletion at, ITPR1, underlies Ataxia in mice and spinocerebellar ataxia 15 in humans[J]. PLoS Genet 3(6):e108. https://doi.org/10.1371/journal.pgen.0030108
Lindquist SG, Mueller LB, Dali CI et al (2017) A novel TTBK2 de novo mutation in a Danish family with early-onset spinocerebellar Ataxia[J]. Cerebellum 16(1):268–271. https://doi.org/10.1007/s12311-016-0786-9
Bauer P, Stevanin G, Beetz C et al (2010) Spinocerebellar ataxia type 11 (SCA11) is an uncommon cause of dominant ataxia among French and German kindreds[J]. J Neurol Neurosurg Psychiatry 81(11):1229–1232. https://doi.org/10.1136/jnnp.2009.202150
Johnson J, Wood N, Giunti P et al (2008) Clinical and genetic analysis of spinocerebellar ataxia type 11[J]. Cerebellum 7(2):159–164. https://doi.org/10.1007/s12311-008-0022-3
Xu Q, Li X, Wang J et al (2010) Spinocerebellar ataxia type 11 in the Chinese Han population[J]. Neurol Sci 31(1):107–109. https://doi.org/10.1007/s10072-009-0129-4
Edener U, Kurth I, Meiner A et al (2009) Missense exchanges in the TTBK2 gene mutated in SCA11[J]. J Neurol 256(11):1856–1859. https://doi.org/10.1007/s00415-009-5209-0
Acknowledgments
The authors wish to thank the patient and her family members for cooperation.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
YD, JF, and YZ performed the acquisition of data and analytical studies, and wrote the manuscript; XQ and MZ conducted and performed the periodic clinical monitoring; XQ planned the study, checked the final form of the manuscript, and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of The Second Affiliated Hospital of Nanchang University. A written informed consent form was obtained from each study participant.
Consent to publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Deng, Y., Fu, J., Zhong, Y. et al. First finding of familial spinal cerebellar Ataxia11 in China: clinical, imaging and genetic features. Neurol Sci 41, 155–160 (2020). https://doi.org/10.1007/s10072-019-04052-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-019-04052-6