
Abstract
Non-dystrophic myotonias are characterized by clinical overlap making it challenging to establish genotype-phenotype correlations. We report clinical and electrophysiological findings in a girl and her father concomitantly harbouring single heterozygous mutations in SCN4A and CLCN1 genes. Functional characterization of N1297S hNav1.4 mutant was performed by patch clamp. The patients displayed a mild phenotype, mostly resembling a sodium channel myotonia. The CLCN1 c.501C>G (p.F167L) mutation has been already described in recessive pedigrees, whereas the SCN4A c.3890A>G (p.N1297S) variation is novel. Patch clamp experiments showed impairment of fast and slow inactivation of the mutated Nav1.4 sodium channel. The present findings suggest that analysis of both SCN4A and CLCN1 genes should be considered in myotonic patients with atypical clinical and neurophysiological features.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Non-dystrophic myotonias (NDM) are rare neuromuscular diseases clinically characterized by muscle stiffness. These include dominant and recessive myotonia congenita (MC; Thomsen disease MIM160800 and Becker disease MIM255700), caused by mutations in the CLCN1 gene encoding the skeletal muscle chloride channel type 1 (ClC-1), as well as paramyotonia congenita (PC; MIM168300) and sodium channel myotonia (SCM; MIM608390), which are both caused by mutations in the SCN4A gene encoding the voltage-gated sodium channel (Nav1.4) alpha subunit [1]. MC is more common than PC and SCM, as confirmed by a recent prevalence study of skeletal muscle channelopathies in the UK [2]. Although some clinical clues and pattern of inheritance point out specific entities, considerable clinical overlap has been reported in NDM, making it difficult to decide on which gene has to be sequenced first [3]. Electromyography (EMG) response to the short and prolonged exercise tests (ET) may help to address the genetic screening and better characterize the phenotype [4,5,6].
We describe here the clinical and electrophysiological findings observed in a girl and her father affected by myotonia and concomitantly harbouring single heterozygous mutations in SCN4A and CLCN1 genes. In addition, we performed patch clamp analysis of the mutant hNav1.4 sodium channel to investigate the effects of the new SCN4A mutation.
Materials and methods
Genetics
Written informed consent for genetic analysis was obtained from the proband, her parents and her brother, as required by the Ethical Committee of the Foundation Neurological Institute Carlo Besta, in accordance with the Helsinki Declaration.
Genomic DNA was extracted from peripheral blood according to standard protocols [7]. The screening for CLCN1 gene mutations was performed as previously described [8, 9]. The genomic structure and intron boundary sequences of SCN4A gene were derived from the Gene Table of Neuromuscular Disorders (http://www.musclegenetable.fr/) and then the GENATLAS database (http://genatlas.medecine.univ-paris5.fr/). Primer sequences specific for the 24 exons were identified using the Primer Express software package (Thermo Fisher Scientific, Monza, Italy) and synthesized by Primm (Milan, Italy). Primer sequences and specific annealing temperature are available on request. The PCR products were purified by EuroSAP (EuroClone, Pero, Italy) and sequenced by bidirectional sequencing using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), on a 3130xlGenetic Analyzer (Thermo Fisher Scientific). The obtained sequences were analyzed with SeqScape v.3.0 software (Thermo Fisher Scientific) and compared with reference wild-type sequence (GenBank accession numbers: NM_000083.2 and NM_000334.4, for CLCN1 and SCN4A, respectively).
Mutagenesis and expression of recombinant sodium channels
The SCN4A p.N1297S mutation was introduced in the pRc/CMV-hNav1.4 vector encoding the human skeletal muscle voltage-gated sodium channel, using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA), and confirmed by complete sequencing. The wild-type (WT) or N1297S channels were co-expressed with the sodium channel β1 subunit and the CD8 receptor gene reporter (pCD8-IRES-hβ1 plasmid), in HEK293 cells, using the calcium-phosphate co-precipitation method, as previously described [10]. Patch clamp was performed on cells spotted by microbeads coated with anti-CD8 antibody (Dynal-Invitrogen, Milan, Italy).
Whole-cell recording and data analysis
Whole-cell sodium currents (I Na) were recorded using Axopatch 1D patch-clamp amplifier (Axon Instruments, Union City, CA) at room temperature (20–22 °C), as previously described [11]. The pipette solution was made of the following (in mM): 120 CsF, 10 CsCl, 10 NaCl, 5 EGTA and 5 Cs-HEPES (pH 7.2). The bath solution was as follows (in mM): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 Na-HEPES and 5 glucose (pH 7.4). Resistance of patch pipettes ranged from 1 to 3 MΩ. Only data obtained in cells displaying series resistance errors < 5 mV were considered for analysis.
Voltage clamp protocols are described in Figs. 2 and 3. Because of the well-known spontaneous shift of sodium channel voltage dependence during whole-cell experiments, the various protocols were applied respecting a constant time sequence in all the cells.
Data analysis was performed using pCLAMP 10.3 (Axon Instruments) and SigmaPlot 8.02 (Systat Software GmbH, Erkrath, Germany). Data and fit parameters are reported as mean ± SEM from n cells. Statistical comparison between WT and N1297S was performed by unpaired Student’s t test, with p < 0.05 considered as significant.
Results
Case report
A woman, now 26 years old, with no apparent family history of neuromuscular disorders, presented at the age of 18 with fatigue and painful stiffness in the upper and lower limbs, mainly in hand and thigh muscles. In addition, she complained of occasional episodes of transient lower limb muscle weakness when standing up after prolonged rest. The symptoms markedly worsened with cold and improved with exercise. At the first neurological examination, performed at 20 years of age, the only clinical sign was percussion-activated myotonia in the hands. Lower limb T1-weighted muscle MRI (1.5 T) was normal. Muscle biopsy showed mild non-specific myopathic signs. Molecular analysis for CNBP and DMPK genes was negative, excluding myotonic dystrophies. Assuming a mild form of dominant myotonia congenita, CLCN1 gene was sequenced, revealing the presence of an already described c.501C>G (p.F167L) mutation in exon 4, usually reported in recessive pedigrees [8, 12, 13]. Last clinical assessment at the age of 24 years showed handgrip myotonia with warm-up phenomenon. Muscle trophism and power were normal, with the exception of a minimal thigh flexion weakness. On EMG, myotonic discharges were observed in all the examined muscles, including tongue, paraspinalis muscles and orbicularis oculi. Despite the reported cold sensitivity, the short ET, performed as previously described [4,5,6], failed to detect any significant changes in compound muscle action potential (CMAP) amplitude/area, both at room temperature and after cooling (Fig. 1a). Also the long ET did not reveal any relevant CMAP modification (Fig. 1c).

Short and long exercise EMG tests. a Repeated short exercise test in patients 1 (a) and 2 (b). Recording of the abductor digiti minimi compound muscle action potential (CMAP) amplitude during three successive short exercises performed at room temperature (black line) and after 7 min of cold exposure (grey line). The first point (100%) corresponds to pre-exercise recording and the subsequent points are related to post-exercise recording during the 50-s resting period and are expressed as percentage of the first value. There was no significant (i.e. < 80 or > 120%) change in CMAP amplitude both at room temperature and after cooling. Long exercise test recording of the abductor digiti minimi compound muscle action potential (CMAP) in patients 1 (c) and 2 (d) performed during 1, 2 and 3 min of exercise (1, 2, 3 days) and 30 min after exercise. No significant (i.e. < 80 or > 120%) CMAP variation in amplitude (black line) or area (grey line) was observed. The first point (T0, 100%) corresponds to pre-exercise recording and the subsequent points are expressed as percentage of the first value
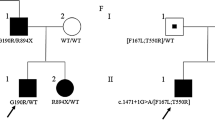
Taken together, clinical and neurophysiological findings showed features somewhat atypical for a dominant MC, including the presence of painful stiffness and transient weakness and the lack of CMAP decrement after exercise, especially with cooling. It should be noted however that the short and long ET are sometimes reported as unreliable in discriminating between SCM and dominant MC [5, 14]. In order to further investigate the case, we screened the SCN4A gene and detected in heterozygous form a mutation, c.3890A>G (p.N1297S) in exon 21, not previously reported or included in the databases (1000 Genomes Project www.1000genomes.org; Exome Variant Server www.evs.gs.washington.edu). Genetic screening was thus extended to the other family members, and the same CLCN1 and SCN4A mutations were found in the 53-year-old father, who had never required neurological attention, even if he complained of mild and painful muscle stiffness in the hands since young age. Father’s neurological examination showed percussion myotonia in the hands and calves and diffuse myotonic discharges on EMG; the ET displayed the same pattern of the affected daughter (Fig. 1b, d). The proband and the father have no eye closure myotonia.
The proband asymptomatic brother and mother did not carry any pathogenic variants.
Functional characterization of N1297S hNav1.4 mutant
Wild-type and N1297S sodium currents recorded in tsA201 cells showed no obvious differences (Fig. 2a–c). The current-voltage relationships of WT and N1297S were well superimposed (Fig. 2d), the voltage dependence of activation being similar (not shown). There was no significant difference in exponential time constants for entry in or recovery from fast inactivation between WT and N1297S (not shown).

Families of sodium currents generated by wild-type and N1297S hNav1.4 channels in transfected HEK293 cells. a The voltage clamp protocol consisted in 25-ms test pulses ranging from − 70 to + 40 mV, applied in 10-mV increments from the holding potential of − 150 mV. b Representative family of N1297S hNav1.4 currents. c Representative family of wild-type hNav1.4 currents. d The current-voltage relationships of N1297S and WT were superimposed
The voltage dependences of fast and slow inactivation were studied using conventional two-pulse voltage protocols (Fig. 3). To measure slow inactivation, a 20-ms interval was introduced before the test pulse to allow recovery from fast inactivation. The normalized peak sodium current amplitude measured during the test pulse was reported as a function of conditioning pulse voltage. The relationships were fit to a Boltzmann equation, I/I max = 1/{1 + exp([V − V f]/K f)} or I/I max = I R1 + (1 − I R1)/{1 + exp([V − Vs]/K s)}, where V f and Vs are the half-maximum voltages for fast and slow inactivation, K f and K s are the slope factors, and I R1 is the residual current. Fit parameter values are reported in Table 1. The N1297S mutation induced a significant positive shift of the half-maximum fast inactivation potential by 7 mV, which is expected to contribute to clinical myotonia (Fig. 3a). The voltage dependence of slow inactivation of N1297S and WT is shown in Fig. 3b. Although the half-maximum inactivation voltage was not significantly altered, the residual current at more positive voltage (I R1) was significantly greater for N1297S compared to WT (Table 1). A similar difference in residual current between WT and N1297S was observed using a protocol aimed at measuring the kinetics for slow inactivation development (Fig. 3c). The relationship was fit to an exponential decay equation, I t/I c = I R2 + (1 − I R2)exp(−t/tau), where I t is the current amplitude during the test pulse, I c is the current amplitude during the conditioning pulse, tau is the exponential time constant, and I R2 is the residual current (Table 1). The time constant to entry in the slow inactivated state was similar for N1297S and WT channels, but fewer N1297S channels entered the slow inactivation state at positive voltages.

Fast and slow inactivation of wild-type and N1297S channels. a To plot fast inactivation voltage dependence, sodium currents measured during a test pulse at − 20 mV were reported as a function of the conditioning pulse ranging from − 150 to − 20 mV, applied in 10-mV increments. The relationships, fitted to a Boltzmann function (equation given in the text), were positively shifted by 7 mV for N1297S compared to WT. b Slow inactivation was induced by 30-s conditioning pulses. Fast inactivation was removed by an intermediate 20-ms pulse at − 160 mV, before assessing channel availability at − 20 mV (inset). A fraction of channels do not enter slow inactivation. The relationships of WT and N1297S channels fitted to a Boltzmann equation (given in the text) show that less N1297S channels enter the slow inactivated state than the WT. c To measure the kinetics of entry in the slow inactivated state, the cells were depolarized to 0 mV for a time lasting 0.01 to 30 s (conditioning pulse), and channel availability was assessed by a test pulse to 0 mV. Again, fast inactivation was removed by an intermediate 20-ms pulse at − 160 mV. The relationships fitted to a monoexponential decay equation show no change in kinetics but a greater proportion of non-inactivated N1297S channels at positive voltages. The fit parameter values and statistical analysis are provided for in Table 1
Discussion
NDM are characterized by clinical overlap making it difficult to establish proper genotype-phenotype correlations. Indeed, SCM and dominant MC may share some clinical features, especially warm-up phenomenon, handgrip myotonia and cold sensitivity [13, 14]. However, cold trigger seems to be more common in SCM, whereas warm-up phenomenon is more frequently associated with dominant MC; handgrip myotonia seems to be equally common in SCM and dominant MC [13]. Some clinical differences exist, including eye closure myotonia and painful stiffness, which are highly suggestive of SCM [14]. Transient weakness is uncommon in dominant MC and more frequently associated with the recessive MC; it has never been reported in SCM [15]. Finally, absence of CMAP modification during ET and cooling is more common in SCM [5, 6]. We report here two affected relatives, each carrying concomitantly two heterozygous mutations, one in the CLCN1 gene and the other in the SCN4A gene, and displaying clinical features overlapping chloride and sodium channel myotonia. Although eye closure myotonia was not observed in our patients, overall clinical phenotype was more suggestive of SCM. Furthermore, the transient weakness reported by the proband was only occasional and we never detected any relevant CMAP decrement upon short ET, which normally represents the neurophysiological correlate of transient weakness, especially in recessive MC.
The F167L mutation in the CLCN1 gene has been previously reported usually in association with other CLCN1 mutations in patients with recessive MC, and its pathological role has been long debated, although it has never been detected in healthy individuals [9, 12, 16,17,18,19,20]. Previous patch clamp examination of p.F167L hClC-1 mutants in transfected HEK293 cells showed little or no difference with wild-type channels [12, 15, 21]. Thus, the clinical phenotype is usually mainly attributed to the dysfunction of the compound mutation.
The SCN4A p.N1297S mutation found in the two affected relatives has never been reported before. In position 1297, the substitution of asparagine (N) with lysine (K) has been previously detected in a severe neonatal non-dystrophic myotonic case, characterized by frequent cold-induced episodes of myotonia and muscle weakness, involving also respiratory muscles with prolonged apnea [22]; in addition, that patient had facial dysmorphisms, muscle hypertrophy and psychomotor retardation and died at the age of 20 months due to respiratory infection. Thus, substitution of N1297 by two different amino acids appears to lead to different symptoms and severity. Serine and lysine residues share similarities in bulk but opposite charges, which may determine different effects on channel behaviour. The physicochemical properties of serine are more similar to those of asparagine than those of lysine, that may explain, at least in part, why N1297S carriers are less severely affected than N1297K carrier. The functional consequence of N1297K on sodium channel behaviour has not been reported. Here we report the functional analysis of N1297S. The mutation induced a 7-mV positive shift in the voltage dependence of fast inactivation, without significantly altering the kinetics of entry in or recovery from fast inactivation. Such a shift is expected to increase the availability of mutant channels, thereby contributing to muscle over-excitability [23, 24]. Alteration of fast inactivation is in agreement with the location of N1297 in the intracellular DIII-DIV linker of the channel protein, which is thought to act as the inactivation gate occluding the mouth of the pore quickly after opening [25]. Other mutations of neighbouring amino acids have been associated with severe myotonia, including G1306E [11, 26]. Importantly, as for N1297, different substitutions of G1306 can lead to symptoms of various severity [23]. In addition, we also observed an alteration of N1297S slow inactivation resulting in an increased proportion of non-inactivated channels after long depolarization. Impaired slow inactivation of sodium channel mutants is thought to be commonly associated with paralytic attacks, which were not reported by our patients [27, 28].
Although clinical and neurophysiological findings found in our patients are not entirely typical of a SCN4A-linked disease, most of them suggests a SCM, thus supporting the pathogenic role of the SCN4A mutation. Nevertheless, previous studies have proposed that SCN4A and CLCN1 mutations may act synergistically to increase the propensity for myotonic discharges, thereby influencing clinical and neurophysiological phenotype [29]. Coexistence of heterozygous SCN4A and CLCN1 mutations in five patients from three myotonic families investigated for both genes due to some atypical features has been recently described [30]. Furthermore, a patient with periodic paralyses and myotonia displayed a combination of SCN4A and CLCN1 simple heterozygous mutations, although clinical phenotype was largely due to the mutation in the Nav1.4 channel [31]. In myotonic dystrophy type 1 or 2, the concomitance of heterozygous CLCN1 or SCN4A mutations produces a more severe myotonia, thus suggesting that different genes involved in myotonic disorders may modify the phenotype and thus contribute to clinical variability [32,33,34,35]. In addition, other factors such as hormones have been described to influence SMC patient phenotype [36]. As already reported, the CLCN1 p.F167L may reduce the severity of symptoms in recessive MC patients carrying other specific CLCN1 mutations [15]. It should be noted here that the patients, especially the father, display a mild phenotype despite the presence of the SCN4A mutation that significantly impairs fast and slow inactivation. There is a possibility that one mutant may mitigate the effects of the other on sarcolemma excitability. Further experiments are warranted to verify whether and how the mutations in the two genes may influence each other.
Taken together, the present findings suggest that analysis of both SCN4A and CLCN1 genes should be considered in myotonic patients with atypical clinical and neurophysiological features.
References
Cannon SC (2015) Channelopathies of skeletal muscle excitability. Compr Physiol 5:761–790
Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC et al (2013) Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 80:1472–1475
Suetterlin K, Männikkö R, Hanna MG (2014) Muscle channelopathies: recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol 27:583–590
Fournier E, Arzel M, Sternberg D, Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B et al (2004) Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol 56:650–661
Fournier E, Viala K, Gervais H, Sternberg D, Arzel-Hézode M, Laforet P et al (2006) Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol 60:356–365
Tan SV, Matthews E, Barber M, Burge GA, Rajakulendran S, Fialho D et al (2011) Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol 69:328–340
Madisen L, Hoar DI, Holroyd CD, Crisp M, Hodes ME (1987) DNA banking: the effects of storage of blood and isolated DNA on the integrity of DNA. Am J Med Genet 27:379–390
Brugnoni R, Galantini S, Confalonieri P, Balestrini MR, Cornelio F, Mantegazza R (1999) Identification of three novel mutations in the major human skeletal muscle chloride channel gene (CLCN1), causing myotonia congenita. Hum Mutat 14:447
Brugnoni R, Kapetis D, Imbrici P, Pessia M, Canioni E, Colleoni L et al (2013) A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet 58:581–587
Desaphy JF, Carbonara R, D'Amico A, Modoni A, Roussel J, Imbrici P et al (2016) Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology 86:2100–2108
Desaphy J-F, De Luca A, Tortorella P, De Vito D, George AL Jr, Conte Camerino D (2001) Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology 57:1849–1857
Zhang J, Bendahhou S, Sanguinetti MC, Ptacek L (2000) Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology 54:937–942
Trivedi JR, Bundy B, Statland J, Salajegheh M, Rayan DR, Venance SL et al (2013) Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain 136:2189–2200
Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D et al (2010) The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 133:9–22
Desaphy JF, Gramegna G, Altamura C, Dinardo MM, Imbrici P, George AL Jr et al (2013a) Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp Neurol 248:530–540
George AL Jr, Sloan-Brown K, Fenichel GM, Mitchell GA, Spiegel R, Pascuzzi RM (1994) Nonsense and missense mutations of the muscle chloride channel gene in patients with myotonia congenita. Hum Mol Genet 3:2071–2072
Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch TJ, Koch MC (1995) Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet 57:1325–1334
Michel P, Sternberg D, Jeannet PY, Dunand M, Thonney F, Kress W et al (2007) Comparative efficacy of repetitive nerve stimulation, exercise, and cold in differentiating myotonic disorders. Muscle Nerve 36:643–650
Modoni A, D’Amico A, Dallapiccola B, Mereu ML, Merlini L, Pagliarani S et al (2011) Low-rate repetitive nerve stimulation protocol in an Italian cohort of patients affected by recessive myotonia congenita. J Clin Neurophysiol 28:39–44
Mazón MJ, Barros F, De la Peña P, Quesada JF, Escudero A, Cobo AM et al (2012) Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul Disord 22:231–243
Lucchiari S, Ulzi G, Magri F, Bucchia M, Corbetta F, Servida M et al (2013) Clinical evaluation and cellular electrophysiology of a recessive CLCN1 patient. J Physiol Pharmacol 64:669–678
Gay S, Dupuis D, Faivre L, Masurel-Paulet A, Labenne M, Colombani M et al (2008) Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am J Med Genet 146A:380–383
Mitrović N, George AL Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F (1995) Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol 487:107–114
Hayward LJ, Brown RH Jr, Cannon SC (1996) Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol 107:559–576
Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N et al (1989) Structural parts involved in activation and inactivation of the sodium channel. Nature 339:597–603
Desaphy J-F, Modoni A, Lo Monaco M, Conte Camerino D (2013b) Dramatic improvement of myotonia permanens with flecainide: a two-case report of a possible bench-to-bedside pharmacogenetics strategy. Eur J Clin Pharmacol 69:1037–1039
Cummins TR, Sigworth FJ (1996) Impaired slow inactivation in mutant sodium channels. Biophys J 71:227–236
Hayward LJ, Brown RH Jr, Cannon SC (1997) Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J 72:1204–1219
Skov M, Riisager A, Fraser JA, Nielsen OB, Pedersen TH (2013) Extracellular magnesium and calcium reduce myotonia in ClC.1 inhibited rat muscle. Neuromuscul Disord 23:489–502
Furby A, Vicart S, Camdessanché JP, Fournier E, Chabrier S, Lagrue E et al (2014) Heterozygous CLCN1 mutations can modulate phenotype in sodium channel myotonia. Neuromuscul Disord 24:953–999
Kato H, Kokunai Y, Dalle C, Kubota T, Madokoro Y, Yuasa H et al (2016) A case of non-dystrophic myotonia with concomitant mutations in the SCN4A and CLCN1 genes. J Neurol Sci 369:254–258
Sun C, Van Ghelue M, Tranebjærg L, Thyssen F, Nilssen Ø, Torbergsen T (2011) Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet 80:574–580
Cardani R, Giagnacovo M, Botta A, Rinaldi F, Morgante A, Udd B et al (2012) Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol 259:2090–2099
Bugiardini E, Rivolta I, Binda A, Soriano Caminero A, Cirillo F, Cinti A et al (2015) SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord 25:301–307
Peddareddygari LR, Grewal AS, Grewal RP (2016) Focal seizures in a patient with myotonic disorder type 2 co-segregating with a chloride voltage-gated channel 1 gene mutation: a case report. J Med Case Rep 10:167
Burge JA, Hanna MG, Schorge S (2013) Nongenomic actions of progesterone and 17beta-estradiol on the chloride conductance of skeletal muscle. Muscle Nerve 48:589–591
Funding
This research was supported by the Italian Telethon Foundation (Grant GGP14096) and the Italian Ministry of Health (Grant GR-2009-1580433).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Written informed consent for genetic analysis was obtained from the proband, her parents and her brother, as required by the Ethical Committee of the Foundation Neurological Institute Carlo Besta, in accordance with the Helsinki Declaration.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Maggi, L., Ravaglia, S., Farinato, A. et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics 18, 219–225 (2017). https://doi.org/10.1007/s10048-017-0525-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-017-0525-5