
Abstract
Since identification of mutations in the ATM gene leading to ataxia-telangiectasia, enormous efforts have been devoted to discovering the roles this protein plays in DNA repair as well as other cellular functions. Even before the identification of ATM mutations, it was clear that other diseases with different genomic loci had very similar neurological symptoms. There has been significant progress in understanding why cancer and immunodeficiency occur in ataxia-telangiectasia even though many details remain to be determined, but the field is no closer to determining why the nervous system requires ATM and other DNA repair genes. Even though rodent disease models have similar DNA repair abnormalities as the human diseases, they have no consistent, robust neuropathological phenotype making it difficult to understand the neurological underpinnings of disease. Therefore, it may be useful to reassess the neurological and neuropathological characteristics of ataxia-telangiectasia in human patients to look for potential commonalities in DNA repair diseases that result in ataxia. In doing so, it is clear that ataxia-telangiectasia and similar diseases share neurological features other than merely ataxia, such as length-dependent motor and sensory neuropathies, and that the neuroanatomical localization for these symptoms is understood. Cells affected in ataxia-telangiectasia and similar diseases are some of the largest single nucleated cells in the body. In addition, a subset of these diseases also has extrapyramidal movements and oculomotor apraxia. These neurological and neuropathological similarities may indicate a common DNA repair related pathogenesis with very large cell size as a critical risk factor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ataxia-telangiectasia (A-T) is an autosomal recessive disease that has long fascinated clinicians and scientists alike (OMIM #208900) [1]. A-T was likely originally described in 1926 [2] and again in 1941 [3]. However, it was not until a series of patients described by Boder and Sedgewick [4] as well as Wells and Shy [5] that A-T became widely appreciated as a clinical syndrome. Although rare, with an estimated incidence of 1 in 40,000 to 100,000 births [6–8], A-T has been of significant interest because of patients’ unusual combination of neurological degeneration, cancer predisposition, immunodeficiency, radiation sensitivity, and telangiectasia (capillary dilatation on the face and sclera most prominently) as well as specific laboratory abnormalities (such as elevated alpha fetal protein levels) that aid in diagnosis. Cancer and diseases of the respiratory system (possibly secondary to immunodeficiency) are the most common causes of death, but neurological degeneration results in significant disability over the majority of a patient’s life [8]. Cells derived from A-T patients were found to be sensitive to ionizing radiation and have chromosome rearrangements, first suggesting ATM’s role in DNA repair and genome stability [9–12]. After the genetic cause was identified, mutations in the gene ataxia-telangiectasia mutated (ATM) [13], a great deal of excellent work has focused on understanding the myriad of functions of this protein. Through this research, there is a reasonable insight into the causes of both immunodeficiencies and cancer [14]. However, there is still no clear mechanistic understanding of neurological degeneration.
In addition to A-T, there are several other DNA repair associated neurological diseases that have extremely similar neurological symptoms. Different symptoms have variable penetrance and severity at different times during the course of disease, but common symptoms suggest or have been demonstrated to have similar neuropathological localization between diseases. The similarities in neurological and neuropathological phenotypes in multiple DNA repair diseases suggest the possibility of a similar neuropathological mechanism(s). Neurologists and neuropathologists who study A-T and similar diseases may have limited understanding of the details of DNA repair. Basic scientists who study DNA repair may not understand the nuances of A-T’s neurological pathology. This is an attempt to partially bridge that gap. Since the goal is to examine neurological aspects of A-T and similar diseases, the neurological symptoms will remain the focus and other clinical phenotypes may be only briefly mentioned.
A-T has been a distinct genetic disease for over 5 decades and is well characterized both neurologically and pathologically so it will be the source for comparison to other diseases. Very similar neurological symptoms are found in other DNA repair diseases including A-T-like disease (MRE11) (OMIM #604391) [15]; ataxia, early onset, with oculomotor apraxia and hypoalbuminemia (EAOH) (a.k.a. ataxia with oculomotor apraxia 1) (APTX) (OMIM #208920) [16, 17]; spinocerebellar ataxia, autosomal recessive 1 (SCAR1) (a.k.a. ataxia with oculomotor apraxia 2) (SETX) (OMIM #606002) [18]; and spinocerebellar ataxia with neuropathy (TDP1) (OMIM #607250) [19], and in some patients with epileptic encephalopathy, early infantile, 10 (a.k.a microcephaly with seizures) (PNKP) (OMIM #605610) [20, 21]. Even though ataxia is not the only neurological symptom, this group of genetic conditions will be referred to as DNA repair ataxia diseases for this phrase’s relative succinctness.
Multiple groups have created mouse models of DNA repair ataxia diseases, including Atm [22–28]. While Atm knockout mice have abnormalities in DNA repair and develop cancer and immunodeficiency, they lack dramatic neuropathology such as neuronal loss with some having subtle or variable neurological phenotypes (reviewed by Lavin [29]). Similarly, murine disease models with mutations in Mre11a [30], Tdp1 [31–33], Aptx [34], and Setx [35] do not have dramatic neuropathological phenotypes similar to the human diseases even though they also replicate the DNA repair abnormalities. The lack of a robust neuropathological phenotype has limited the ability to test hypotheses concerning mechanisms for neurodegeneration. This emphasizes the importance of understanding the human neurological pathology.
Neurological features of A-T
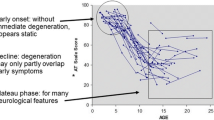
The neurological symptoms and neuropathology will be discussed as a way of illuminating the specific requirements for ATM and other genes in the nervous system. The correlations between a neurological phenotype and loss of specific cell types are typically quite strong for the symptoms discussed. The pathogenesis in Purkinje versus granule cells in the cerebellum will be detailed, but one cannot be certain in which cell type the defect resides since animal models with neurological phenotypes are needed to make definitive determinations. Because of their unknown clinical significance, neuropathological features not clearly associated with A-T’s specific neurological deficits as well as inconsistent or rare pathological findings will not be discussed in detail. Childhood neurological diseases are frequently categorized as degenerative, a function that was previously present but lost, or developmental, deficient development of a function. DNA repair ataxia diseases are degenerative as patients lose functions they previously possessed. The age of onset of neurological symptoms in A-T seems to be affected by mutation severity and can be separated into two categories, early childhood and mid-childhood [36–38]. While later onset neurological symptoms are different in terms of timing and severity, both have the same neuropathological features [38, 39] and early onset neurological disease will be the focus. Many neurological diseases that typically start in childhood, such as mitochondrial or metabolite processing diseases, frequently have milder cases that present in a patient’s 30’s, 40’s, or even later in life. However, A-T neurological symptoms seem to always start prior to adulthood.
In early onset A-T, ataxia and cerebellar degeneration, a particular form of lack of motor coordination, is typically the initial symptom and is found in essentially all patients at some point [8]. Cerebellar atrophy is common but not universal in genetic ataxias. Neurological symptoms in A-T generally start with ataxia, although other symptoms such as extrapyramidal symptoms can predominate at first in later onset disease and weakness can rarely be the earliest symptom [40, 38]. Although not proven, Purkinje cell loss is likely to be the primary cause of cerebellar atrophy and is a universal finding in A-T. Granule cells proliferate, migrate, and synapse to Purkinje cells starting in utero and continuing the first year or two of life when A-T ataxia can start in early onset cases, suggesting that granule cells may be primarily defective (Fig. 1). However, Purkinje cell loss is universal and granule cell loss is not as severe and occasionally not apparent, making ATM more likely required in Purkinje cells [41, 42]. In addition, mouse studies suggest that intrinsic loss of granule cells does not seem to result in severe Purkinje cell losses [43, 44]. Therefore, while it is not definitive, evidence supports Purkinje cells as primarily affected in the cerebellum. Dysarthria or slurred speech is common in A-T. Dysarthria is often difficult to localize neuroanatomically because it can have many different causes. The dysarthria associated with A-T is similar to other forms of ataxia and may be cerebellar in origin [45]. However, drooling is also common in A-T but not a universal symptom in cerebellar ataxias, so involvement of brainstem nuclei controlling the oral pharynx is possible but not clear [8].

Diagram of cerebellum and development. a The mature cerebellar cortex has three layers. Adjacent to the pial surface (outer surface) is the molecular layer (light pink/blue) containing Purkinje cell dendrites, granule cell axons, and a few sparse cells not depicted. Next is a single cell layer of large Purkinje cell bodies (red) followed by the highly abundant granule cell bodies (blue). b Purkinje cells are “born” significantly before granule cell proliferation begins. Granule cells migrate across the pial surface to cover the developing cerebellum forming the external granule cell layer. Granule cell precursors divide (yellow), start to differentiate (green), and when mature (blue) migrate into the cerebellum. As the granule cells migrate through the molecular layer, they extend their axons and form synapses on Purkinje cell dendrites. Granule cell bodies migrate past the Purkinje layer and into the internal granule cell layer with their axons trailing behind. c A cartoon underrepresenting the elaborate arborization of a human Purkinje cell dendrite. The dendrite is planar and viewed here sagittally. d Two Purkinje cells (turned 80° from view seen in c) with two granule cell parallel fibers (axons) extend in the horizontal plane making synapses on multiple Purkinje cell dendrites. Granule cell synapses are required to form the very large Purkinje cell dendritic tree. Therefore, granule cell proliferation is associated with dramatic increases in Purkinje volume via dendritic growth (Color figure online)
In early onset A-T, weakness from loss of motor neurons generally starts to become an issue later in childhood (Fig. 2, left side). In A-T, neuropathy is consistent with loss of axons in the nerve (as opposed to loss of myelin that covers axons) as well as the motor neuron cell bodies within the spinal cord, most pronounced within the lumbar regions (lower spinal cord that innervates the legs) [46–49, 39]. The neuropathy is most severe for the longest axons with distal muscle wasting of the feet and hands common late in disease. Muscle loss is consistent with loss of neurons as opposed to a primary defect in muscle fibers based upon muscle histological pattern, group atrophy [50, 39]. The length-dependent nature of the neuropathy is illustrated by a patient who cannot move his toes and barely his ankles but has normal or near normal strength at shoulders and hips [51].

Illustration of the relative vulnerability of motor and sensory neurons in A-T. The thin black lines in midline represent the spinal cord. On the left are motor neurons with cell bodies residing within the spinal cord whose axons extend to muscles. The motor neurons with the longest axons (red) are most affected in A-T while the motor neurons with shorter axons (green) are less affected. On the right are sensory neurons with cell bodies just outside of spinal cord in clusters, the dorsal root ganglia. A single axon extends out into the periphery as well as into the spinal cord. The most vulnerable neurons have large diameter cell bodies and axons and a single large caliber axon that carries vibration sense or position information (red) from the legs to the brain stem. Sensory neurons with small cell bodies and smaller axonal caliber carry pain or temperature sensation (blue), convey information from the periphery, synapse in the spinal cord at the approximate level they enter (not in the brain stem), and are less affected in either the upper or lower extremities of A-T patients compared to vibration and proprioceptive neurons (Color figure online)
A-T patients lose sensation from loss of sensory neurons (Fig. 2, right side), particularly position and vibration as well as losing deep tendon reflexes, but have relative sparing of cold and pain sensation [45, 49]. Like motor neurons, the neuropathy is axonal and also includes loss of sensory neuronal cell bodies in dorsal root ganglia [50, 46, 8, 49, 39]. The spinal cord shows a striking loss of sensory axons carrying position and vibration sense within the posterior portion of the spinal cord from the leg regions with arm regions relatively intact [50, 45, 39]. The axons that convey joint position and vibration are larger in diameter than those that carry pain and temperature, and the large caliber fibers are lost in A-T nerves [47]. Taken together, this shows that neurons are lost in a length-dependent (longer > shorter), as well in an axonal diameter-dependent (larger > smaller), manner [39]. The loss of deep tendon reflexes in A-T patients is likely due to the loss of sensory neurons, motor neurons, or both [8].
Many A-T patients have prominent movement disorders that appear extrapyramidal in nature [2, 5, 8, 52]. A-T patients’ extrapyramidal symptoms frequently include abnormal posture, tremor (shaking), myoclonus (rapid single jerks of a limb), and/or choreoathetoid movements (slow dancing and/or writhing-like) [8, 52]. Extrapyramidal symptoms are often associated with injury to the basal ganglia but can also come from injury to regions of the thalamus or brainstem such as the substantia nigra. In early onset A-T, extrapyramidal symptoms are prominent later in the course of the disease but at times can predominate to the extent that two early descriptions of A-T described the disease as extrapyramidal as opposed to ataxic in nature [2, 5]. Only a few neuropathological studies have found any potential pathology in the basal ganglia while others found abnormalities in the locus coeruleus and the substantia nigra [53–55, 48, 45, 56, 39]. A-T patients have metabolic changes in their basal ganglia indicative of dysfunction even though the neuroanatomical localization of the extrapyramidal neuropathology remains to be definitely determined [57].
A-T patients can have peculiar eye movements known as oculomotor apraxia that are distinct from those usually found in other ataxic diseases [58, 8]. Oculomotor apraxia is a rare condition where patients have the ability to move eyes in all directions when tracking (following) an object but cannot voluntarily move their eyes to a different location, as when looking back and forth between two different objects [59]. The neuroanatomical localization for oculomotor apraxia is not known in A-T or any other condition [59].
While affecting multiple different types of neurons, A-T does not appear to be a universally progressive neurological disease. As noted above, A-T is a degenerative disease and several different cell types are lost, but the vast majority of the neurons in the brain appear unaffected in A-T. For instance, patients do not seem to have significant cognitive degeneration during the course of their disease. Early impressions were that intellectual disability was present in A-T, but much of that was due to slowness in responses secondary to motor dysfunction [60, 48, 45]. When followed over time, A-T patients can stop acquiring intellectual gains as opposed to losing intellectual capacity that typically occurs during cerebral cortical degeneration. In addition, there is little definitive structural MRI or pathological change in the cerebral cortex, thalamus, or basal ganglia neurons or axons, although it is possible that subtle changes may have not yet been consistently identified. Abnormalities in small blood vessels are present in the brain, but they seem to occur later in disease and are not necessarily correlated with neurodegeneration, so their contribution to neurodegeneration is uncertain [48, 8, 61]. A-T patients have cells with hyperchromatic nuclei with bizarre shapes, but these abnormalities are found in many organs with no correlation to tissue pathology and their clinical significance remains undetermined [46, 62, 55, 48].
Neurological features of other DNA repair ataxia diseases
The other DNA repair ataxia diseases were recognized more recently. Therefore, the clinical and neuropathological literature is not as extensive. Neurological similarities to A-T will only be summarized and neuropathological similarities when known are not detailed due to space constraints (Table 1). Mutations in MRE11A can produce ataxia-telangiectasia-like disorder (ATLD) with degenerative cerebellar ataxia, dysarthria, drooling, oculomotor apraxia, loss of sensation, loss of reflexes, weakness with distal muscle wasting, and extrapyramidal symptoms [63, 64, 15, 65, 66]. Mutations in the aprataxin (APTX) gene lead to ataxia, early onset, with oculomotor apraxia and hypoalbuminemia (EAOH) (a.k.a. ataxia with oculomotor apraxia I, AOA1) with degenerative cerebellar ataxia, dysarthria, extrapyramidal abnormalities, oculomotor apraxia, loss of position and vibration sense, and length-dependent weakness with muscle wasting of the distal extremities [67, 16, 17, 68–75]. Senataxin (SETX) mutations can lead to a disease called spinocerebellar ataxia, autosomal recessive 1 (SCAR1) (a.k.a. ataxia with oculomotor apraxia 2, AOA2) with degenerative cerebellar ataxia, extrapyramidal abnormalities, loss of reflexes, motor and sensory neuropathy particularly affecting vibration and position sense, oculomotor apraxia, some evidence of loss innervation to the spinal motor neurons from the cerebral cortex (a.k.a. upper motor neuron signs), and dysarthria [76, 18, 77–83]. Mutations in tyrosyl-DNA phosphodiesterase 1 (TDP1) result in spinocerebellar ataxia, autosomal recessive with axonal neuropathy (SCAN1) with cerebellar degenerative ataxia, distal muscle weakness, length-dependent loss of vibration and position sense, dysarthria, and loss of reflexes [19]. Patients with mutations in polynucleotide kinase 3-prime phosphatase (PNKP) were first reported with microcephaly, developmental delay, and seizures, called epileptic encephalopathy, early infantile 10 (EEEI 10, a.k.a. microcephaly with seizures) [20]. Subsequently, it was found that some patients in later childhood developed cerebellar degenerative ataxia, as well as sensory and motor neuropathy [21]. Mutations in the DNA repair gene TDP2 lead to intellectual disability, epilepsy, and ataxia, but this very recently identified condition requires more clinical characterization before it is clear how patients with TDP2 mutations may relate neuropathologically to the other DNA repair ataxia diseases [84]. Also currently not included is a single family with a compound homozygous polymorphism in phosphoinositide-3-kinase, regulatory subunit 5 (PIK3R5) (OMIM# 615217) because even though they neurologically appear very similar to other DNA repair ataxia diseases, evidence for PIK3R5 dysfunction in the patients is not yet definitive [85].
In summary, patients with A-T (ATM), ATLD (MRE11A), EAOH (AOA1) (APTX), SCAR1 (AOA2) (SETX), and SCAN1 (TDP1) and some with EEEI10 (PNKP) have nearly identical neurological symptoms including prominent cerebellar ataxia, dysarthria (likely secondary to cerebellar degeneration), length-dependent motor neuropathy, and length-dependent sensory axonal neuropathy of vibration and position sense with loss of reflexes likely secondary to neuropathy (Table 1). Mutations in ATM, MRE11A, APTX, and SETX can lead to prominent extrapyramidal symptoms and oculomotor apraxia. Ataxia is consistent with cerebellar degeneration most likely secondary to loss of Purkinje cells. Weakness is predominately due to loss of motor neurons as opposed to loss of myelination or muscle disease. Sensory changes, predominately position and vibration, are also likely secondary to loss of sensory neurons/axons and not an abnormality of myelin. Loss of reflexes is likely secondary to loss of sensory neurons, although loss of motor neurons may also contribute. There is length-dependent axonal loss of the largest diameter axons. The neuropathological localizations for the extrapyramidal symptoms and oculomotor apraxia are not well understood, although one might suspect that they are similar as these symptoms are quite rare in other degenerative ataxia diseases. In addition, it is also important to note the rather restricted and very specific neurons affected in these diseases as A-T and similar diseases do not appear to have widespread neuronal degeneration.
The neurological symptoms associated with DNA repair ataxia diseases are distinct from other ataxia diseases. Autosomal recessive cerebellar degeneration with both motor and sensory neuropathies is not common but present in all DNA repair ataxia diseases. In autosomal recessive ataxias that do have neuropathy, some are demyelinative while A-T and similar diseases have axonal neuropathies [86]. The most common genetic ataxia, Friedreich’s ataxia (OMIM #229300), has some clinical similarities, but several key differences, generally making this disease reasonably easy to clinically distinguish from DNA repair ataxia diseases [87]. Friedreich’s ataxia is often characterized by ataxia and neuropathy similar to A-T, but neuroimaging of the cerebellum is normal early in disease [88, 86]. In Friedreich’s, some of the ataxia may be mediated via loss of the sensory input to the cerebellum via the spinocerebellar tract as well as prominent loss of the cerebellar dentate neurons (as opposed to Purkinje cells) [89]. Additional features of Friedreich’s ataxia include some overlapping features with DNA repair abnormalities associated with ataxia including dysarthria, sensory neuropathy with loss of reflexes, and length-dependent weakness. However, unlike A-T, Friedreich’s ataxia often has evidence of loss of the connection from cerebral cortex to the spinal cord motor neurons (a.k.a. upper motor neuron signs) early, and scoliosis is prominent while some have bladder disturbances, optic atrophy, and hearing loss. Extraneurological symptoms include frequent and potentially lethal cardiac abnormalities and diabetes mellitus [88, 90]. Friedrich’s ataxia is more likely to be confused clinically with a different disease, ataxia with vitamin E deficiency (TTPA) (OMIM #277460), because of overlapping neurological features [91–93]. Dominant ataxias, sometimes called spinocerebellar ataxias (SCA), have nearly 40 different genetic loci. Some have neuropathy and some have extrapyramidal movements, but it would be rare that they would be clinically confused with DNA repair ataxia diseases because of the combination of neurological symptoms and age of onset [94, 95]. When all neurological symptoms are considered, the neurological similarities between DNA repair ataxia diseases are striking when compared to other ataxia diseases.
DNA repair activities
This is not intended to be a comprehensive review of biochemical roles and mechanisms of DNA repair proteins as many excellent reviews have been previously written [96–99, 14, 100–108]. However, a brief elaboration of DNA repair activities is required to establish some context. ATM is a serine/threonine protein kinase that plays a very central role in many aspects of DNA repair signaling, particularly well studied in double-strand break repair. For example, ATM and its paralog ATR phosphorylate over 700 different proteins after ionizing radiation [109]. In addition to ATM’s role in double-strand break repair, it has been implicated in single-strand break repair, response to hypoxia, oxidative stress, insulin signaling, mitochondrial activity, histone acetylation, and other functions (recently reviewed in Hoche et al. [104] and Shiloh and Ziv [107]). MRE11 is required for optimal activation of ATM under multiple stressors (upstream of ATM activation), is phosphorylated by ATM, and has ATM-independent roles as well [98].
Unlike ATM and MRE11, the other DNA repair ataxia disease proteins seem to have much more simple roles in DNA repair, likely having direct enzymatic repair roles on DNA itself. ATPX, TDP1, and PNKP can interact with single-strand break repair machinery [96, 97, 102, 100]. APTX can remove failed ligase products and other abnormalities that are covalently attached to DNA [34, 110]. TDP1 can help remove arrested topoisomerase I covalently bound to DNA and can process damaged bases during single-strand break repair [111–115]. PNKP can interact with multiple DNA repair pathways in its role as a DNA 3′ phosphate and 5′ kinase [116–118, 20, 119]. SETX is a putative RNA/DNA helicase that may repair DNA damage associated with transcription [120–124]. It is certainly possible that ATM and MRE11 control broad/homeostatic signaling pathways during DNA repair or even have a critical non-DNA repair related role required in neurons. However, APTX, SETX, TDP1, and PNKP are not directly involved in signaling and likely have limited, direct enzymatic repair activities on DNA itself, and it would be surprising that non-DNA repair functions would be discovered for all four proteins. Therefore, since DNA repair ataxia diseases share similar neurological phenotypes, it is reasonable to postulate a common DNA repair pathway/mechanism between these various genes in neurons. Recent evidence of a biochemical relationship between ATM and TDP1 supports the possibility of critical shared DNA repair pathways [125].
Potential stressors that require DNA repair
Understanding the underlying pathological mechanisms can be critical for developing therapeutic interventions in any disease. Although the reasons for differential susceptibility of specific neuronal populations remain elusive in many neurological diseases, it may be useful to consider why specific cells are preferentially lost in DNA repair ataxia diseases. As noted, it is reasonable to propose that DNA repair ataxia diseases result from abnormalities in DNA repair. Currently, there is no evidence that the DNA repair pathways in neurons are significantly different than other cells, and patterns of gene expression have yet to provide insight. Alternatively, it is possible that all cells have similar levels of DNA damage and the limited types of affected neurons are more sensitive to DNA damage than other cells, but evidence for that hypothesis is currently lacking. Therefore, it is possible that the affected cells have more DNA damage. Shared characteristics of affected neurons might suggest “stressors” that lead to DNA damage that requires repair.
Purkinje cells, motor neurons, and sensory neurons are “born,” meaning that they complete their last cell division and become terminally differentiated in utero, years prior to becoming clinically dysfunctional. Therefore, if defects are intrinsic to the dying cells, any required DNA repair activities are more likely to be associated with G1 (sometimes referred to as G0 in neurons since they are incapable of becoming proliferative) and not activities specific to S, G2, and M phases. DNA damage secondary to reactive oxygen species is a commonly proposed mechanism for neurodegeneration in A-T and similar diseases. However, there are several reasons to question this hypothesis as the exclusive source of DNA damage. Cardiac muscle and kidney are not affected in DNA repair ataxia diseases even though they use nearly two times the energy by weight as the brain (brain ~240 kcal/kg/d, heart and kidney ~440 kcal/kg/d) [126]. In addition, the brain does not appear to accumulate more somatic DNA damage than other organs [127, 128], although other studies contradict that assertion [129, 130]. It is also not obvious why only a few cell types appear to be lost in DNA repair ataxia diseases since DNA damage secondary to oxidative damage would presumably be widespread. Finally, the hypothesis that reactive oxygen causes accumulation of DNA damage has never been proven due to the difficulty of altering the amount of reactive oxygen generated or present (via anti-oxidant therapy). Therefore, it may be fruitful to explore additional explanations of the nervous system’s requirements for ATM and other ataxia-associated DNA repair proteins.
Clinical features of motor and sensory neurons indicate that large cellular volume is correlated with vulnerability in DNA repair ataxia diseases. The longest motor and sensory neurons as well as the largest caliber sensory neurons (motor neuron axons are more uniform in diameter) are the most severely affected. In addition, huge dendrites of Purkinje cells (Fig. 1c) that grow in the first few years of life are their distinguishing and unique characteristic, but it is very difficult to estimate their volume. Motor and sensory neurons are likely the largest single nucleated cells in the body, but how much bigger they are than other cells in the body can be unappreciated. For instance, the longest motor neurons are >10,000 times larger than an average hepatocyte. The volume of a cell body that is roughly spherical can be calculated as a sphere and an axon a cylinder. A hepatocyte has an approximate diameter of 20 μm and volume of 4.2 × 10−9 cm3. A motor neuron cell body can have a diameter of 50 μm and volume of 65 × 10−9 cm3, not including the dendrites. In adults, axons that extend to the foot can be 1 m long (or greater) and can have a diameter of up to 20 μm and volume of 310,000 × 10−9 cm3. This shows that motor neurons are orders of magnitude larger than other cell types, and the vast majority of the volume of a motor neuron is within the axon. The lack of clear neuropathology in murine models of DNA repair ataxia diseases may be explained by the much smaller sizes of motor and DRG neurons as well as Purkinje cells in mice relative to humans.
The extreme growth in volume of motor and sensory neurons and other vulnerable cells may cause unique stressors for these very large, single nucleated cells. DNA repair ataxia diseases’ neurological symptoms develop in childhood at the time the affected cells need to grow to achieve their very large sizes. While these cells require proportionally larger amounts of amino acids, energy (glucose and fatty acids), etc., absorbing these building blocks may not be limiting because of these cells’ proportionally large surface area. Energy production could be an issue as mitochondria along with their intrinsic mitochondrial genome would presumably be required in proportionally very large numbers. Critically, mitochondria are produced via fission (each mitochondria can divide forming two mitochondria) so their growth can be logarithmic. Defects in mitochondria proliferation dynamics, particularly producing a sufficient number of mitochondria or maintaining them, could be important and have been implicated in multiple neurodegenerative diseases [131, 132] while ATM has been shown to play a role in mitochondrial function [133–136]. An additional potential stressor for producing these huge volumes may be the increase in transcriptional requirements. Most tissues grow dramatically in size via cell proliferation. Every cell division doubles the number of transcriptional units (genomes) and allows for growth in volume of a tissue. Very large neurons must achieve their enormous sizes with the same complement of DNA found in every cell with increases in transcription and translation. Not surprisingly, large cells have large nuclei and nucleoli (sources of rRNA transcription and ribosomes) as well as high rates of transcription, making transcription a potential source of DNA damage [137, 138].
In summary, A-T neurological features have potentially important lessons for understanding the requirements for ATM-related neuropathology. Both neurological and neuropathological studies suggest that the localization of neuronal defects in A-T is restricted to a reasonably small number of cell types, including cerebellar Purkinje cells, motor and sensory neurons, and likely some others that have not been definitively determined. The other DNA repair ataxia diseases seem to have very similar clinical phenotypes both in terms of neurological features and neuropathology including cerebellar ataxia and motor and sensory neuropathy (position and vibration) as well as some having extrapyramidal movements and peculiar eye movements distinguishing these diseases from other forms of ataxia. The size of neurons, particularly during their periods of cell volume growth, may be a critical risk factor for neuronal susceptibility in DNA repair ataxia diseases. This susceptibility is illustrated by the size of cells lost, particularly motor and sensory neurons via the length-dependent nature of the neuropathies. In addition, neurological symptoms present as neurons are growing in volume during childhood or early teen years and not later in adulthood after cell volume increases have ceased. Purkinje cells are often affected when their huge dendrites are growing as granule cells are forming synapses. Size as a risk factor may also explain why rodent disease models do not display neuropathological changes found in the much larger human cells. The DNA repair ataxia disease gene proteins may eventually be tied to a single cellular insult such as oxidative damage, mitochondrial production/repair, transcription-related damage, or other processes. Understanding the particular vulnerabilities in the neurons lost in DNA repair ataxia diseases may be critical to developing potential therapies as well as providing insight into other neurological diseases affecting very large neurons.
References
Online Mendelian Inheritance in Man, OMIM. (2013) Johns Hopkins University (Baltimore, MD). http://omim.org/
Syllaba L, Henner K (1926) Contribution à l'étude de l'indépendance de l'athétose double idiopathique et congénitale. Atteinte familiale, syndrome dystrophique, signe du résau vasculaire conjonctival, intégrité psychique. Rev Neurol 1:541–562
Louis-Bar D (1941) Sur un syndrome progressif cormprenant des télangiectasies capillaires cutanées et conjonctivales symétriques, à disposition naevoïde et des troubles cérébelleux. Confinia Neurologica 4:32–42
Boder E, Sedgwick RP (1958) Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 21(4):526–554
Wells CE, Shy GM (1957) Progressive familial choreoathetosis with cutaneous telangiectasia. J Neurol Neurosurg Psychiatry 20(2):98–104
Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT (1986) The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet 39(5):573–583
Pippard EC, Hall AJ, Barker DJ, Bridges BA (1988) Cancer in homozygotes and heterozygotes of ataxia-telangiectasia and xeroderma pigmentosum in Britain. Cancer Res 48(10):2929–2932
Sedgwick RP, Boder E (1991) Ataxia-Telangiectasia. In: Vinken PJ, Bruyn GW, Klawans HL, Vianney de Jong JMB (eds) Hereditary Neuropathies and Spinocerebellar Atrophies, vol. 60. Handbook of Clinical Neurology. Elsevier Science Publishing Co. Inc, New York, pp 347–426
Hecht F, Koler RD, Rigas D, Dahnke GS, Case MP, Tisdale V, Miller RW (1966) Leukaemeia and lymphocytes in ataxia-telangiectasia. Lancet 288(7474):1193
Gotoff SP, Amirmokri E, Liebner EJ (1967) Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am J Dis Child 114(6):617–625
Morgan JL, Holcomb TM, Morrissey RW (1968) Radiation reaction in ataxia telangiectasia. Am J Dis Child 116(5):557–558
Oxford JM, Harnden DG, Parrington JM, Delhanty JD (1975) Specific chromosome aberrations in ataxia telangiectasia. J Med Genet 12(3):251–262
Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S et al (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268(5218):1749–1753
de Miranda NF, Bjorkman A, Pan-Hammarstrom Q (2011) DNA repair: the link between primary immunodeficiency and cancer. Ann N Y Acad Sci 1246:50–63. doi:10.1111/j.1749-6632.2011.06322.x
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99(6):577–587
Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonça P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M (2001) The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29(2):189–193. doi:10.1038/ng1001-189
Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S (2001) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29(2):184–188. doi:10.1038/ng1001-184
Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE, M'Zahem A, Dunne E, Bomont P, Benhassine T, Bouslam N, Stevanin G, Brice A, Guimaraes J, Mendonca P, Barbot C, Coutinho P, Sequeiros J, Durr A, Warter JM, Koenig M (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 36(3):225–227
Takashima H, Boerkoel C, John J, Saifi G, Salih M, Armstrong D, Mao Y, Quiocho F, Roa B, Nakagawa M, Stockton D, Lupski J (2002) Mutation of TDP1, encoding a topoisomerase I–dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32(2):267–272. doi:10.1038/ng987
Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, Ganesh VS, Chang BS, Grix A, Hill RS, Topcu M, Caldecott KW, Barkovich AJ, Walsh CA (2010) Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 42(3):245–249. doi:10.1038/ng.526
Poulton C, Oegema R, Heijsman D, Hoogeboom J, Schot R, Stroink H, Willemsen MA, Verheijen FW, van de Spek P, Kremer A, Mancini GM (2013) Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics 14(1):43–51. doi:10.1007/s10048-012-0351-8
Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86(1):159–171
Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P (1996) Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 93(23):13084–13089
Xu Y, Baltimore D (1996) Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10(19):2401–2410
Kuljis RO, Xu Y, Aguila MC, Baltimore D (1997) Degeneration of neurons, synapses, and neuropil and glial activation in a murine Atm knockout model of ataxia-telangiectasia. Proc Natl Acad Sci U S A 94(23):12688–12693
Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ (1998) Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280(5366):1089–1091
Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y (2000) Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A 97(7):3336–3341. doi:10.1073/pnas.050584897
Spring K, Cross S, Li C, Watters D, Ben-Senior L, Waring P, Ahangari F, Lu SL, Chen P, Misko I, Paterson C, Kay G, Smorodinsky NI, Shiloh Y, Lavin MF (2001) Atm knock-in mice harboring an in-frame deletion corresponding to the human ATM 7636del9 common mutation exhibit a variant phenotype. Cancer Res 61(11):4561–4568
Lavin MF (2013) The appropriateness of the mouse model for ataxia-telangiectasia: Neurological defects but no neurodegeneration. DNA Repair (Amst) 12(8):612–619. doi:10.1016/j.dnarep.2013.04.014
Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JH, McKinnon PJ (2009) Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev 23(2):171–180. doi:10.1101/gad.1746609
Katyal S, El-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ (2007) TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J 26(22):4720–4731. doi:10.1038/sj.emboj.7601869
Hirano M, Yamamoto A, Mori T, Lan L, Iwamoto TA, Aoki M, Shimada K, Furiya Y, Kariya S, Asai H, Yasui A, Nishiwaki T, Imoto K, Kobayashi N, Kiriyama T, Nagata T, Konishi N, Itoyama Y, Ueno S (2007) DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann Neurol 61(2):162–174. doi:10.1002/ana.21078
Hawkins AJ, Subler MA, Akopiants K, Wiley JL, Taylor SM, Rice AC, Windle JJ, Valerie K, Povirk LF (2009) In vitro complementation of Tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation. DNA Repair (Amst) 8(5):654–663. doi:10.1016/j.dnarep.2008.12.012
Ahel I, Rass U, El-Khamisy S, Katyal S, Clements P, Mckinnon P, Caldecott K, West S (2006) The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443(7112):713–716. doi:10.1038/nature05164
Becherel OJ, Yeo AJ, Stellati A, Heng EY, Luff J, Suraweera AM, Woods R, Fleming J, Carrie D, McKinney K, Xu X, Deng C, Lavin MF (2013) Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet 9(4):e1003435. doi:10.1371/journal.pgen.1003435
McConville CM, Stankovic T, Byrd PJ, McGuire GM, Yao QY, Lennox GG, Taylor MR (1996) Mutations associated with variant phenotypes in ataxia-telangiectasia. Am J Hum Genet 59(2):320–330
Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, Haites N, Byrd PJ, Taylor AM (1998) ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet 62(2):334–345. doi:10.1086/301706
Verhagen MM, Abdo WF, Willemsen MA, Hogervorst FB, Smeets DF, Hiel JA, Brunt ER, van Rijn MA, Majoor Krakauer D, Oldenburg RA, Broeks A, Last JI, van't Veer LJ, Tijssen MA, Dubois AM, Kremer HP, Weemaes CM, Taylor AM, van Deuren M (2009) Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology 73(6):430–437. doi:10.1212/WNL.0b013e3181af33bd
Verhagen MM, Martin JJ, van Deuren M, Ceuterick-de Groote C, Weemaes CM, Kremer BH, Taylor MA, Willemsen MA, Lammens M (2012) Neuropathology in classical and variant ataxia-telangiectasia. Neuropathol: Off J Jpn Soc Neuropathol 32(3):234–244. doi:10.1111/j.1440-1789.2011.01263.x
Larnaout A, Belal S, Ben Hamida C, Ben Hamida M, Hentati F (1998) Atypical ataxia telangiectasia with early childhood lower motor neuron degeneration: a clinicopathological observation in three siblings. J Neurol 245(4):231–235
Paula-Barbosa MM, Ruela C, Tavares MA, Pontes C, Saraiva A, Cruz C (1983) Cerebellar cortex ultrastructure in ataxia-telangiectasia. Ann Neurol 13(3):297–302. doi:10.1002/ana.410130312
Vinters HV, Gatti RA, Rakic P (1985) Sequence of cellular events in cerebellar ontogney relevent to expression of neuronal abnormalities in ataxia-telangiectasia. In: Gatti RA, Swift M (eds) Ataxia-Telangiectasia Genetics, Neuropathology, and Immunology of a Degenerative Disease of Childhood. Alan R. Liss, Inc., New York, pp 233–255
Rakic P, Sidman RL (1973) Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol 152(2):103–132. doi:10.1002/cne.901520202
Goldowitz D, Mullen RJ (1982) Granule cell as a site of gene action in the weaver mouse cerebellum: evidence from heterozygous mutant chimeras. J Neurosci 2(10):1474–1485
Boder E (1985) Ataxia-Telangiectasia: An Overview. In: Gatti RA, Swift M (eds) Ataxia-telangiectasia: genetics, neuropathology, and immunology of a degenerative disease of childhood. Alan R. Liss, Inc., New York, pp 1–63
Aguilar MJ, Kamoshita S, Landing BH, Boder E, Sedgwick RP (1968) Pathological observations in ataxia-telangiectasia. A report of five cases. J Neuropathol Exp Neurol 27(4):659–676
Martinez AC, Barrio M, Gutierrez AM, Lopez (1977) Abnormalities in sensory and mixed evoked potentials in ataxia-telangiectasia. J Neurol Neurosurg Psychiatry 40(1):44–49
Amromin GD, Boder E, Teplitz R (1979) Ataxia-telangiectasia with a 32 year survival. A clinicopathological report. J Neuropathol Exp Neurol 38(6):621–643
Verhagen MM, van Alfen N, Pillen S, Weemaes CM, Yntema JL, Hiel JA, Ter Laak H, van Deuren M, Broeks A, Willemsen MA (2007) Neuromuscular abnormalities in ataxia telangiectasia: a clinical, electrophysiological and muscle ultrasound study. Neuropediatrics 38(3):117–121. doi:10.1055/s-2007-985899
Strich SJ (1966) Pathological findings in three cases of ataxia-telangiectasia. J Neurol Neurosurg Psychiatry 29:489–499
Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 2–1987. A 30-year-old man with ataxia-telangiectasia and dysphagia (1987). The New England journal of medicine 316 (2):91–100. doi:10.1056/NEJM198701083160206
Shaikh AG, Zee DS, Mandir AS, Lederman HM, Crawford TO (2013) Disorders of Upper Limb Movements in Ataxia-Telangiectasia. PLoS ONE 8(6):e67042. doi:10.1371/journal.pone.0067042
Centerwall WR, Miller MM (1958) Ataxia, telangiectasia, and sinopulmonary infections; a syndrome of slowly progressive deterioration in childhood. AMA J Dis Child 95(4):385–396
De Leon GA, Grover WD, Huff DS (1976) Neuropathologic changes in ataxia-telangiectasia. Neurology 26(10):947–951
Agamanolis DP, Greenstein JI (1979) Ataxia-telangiectasia. Report of a case with Lewy bodies and vascular abnormalities within cerebral tissue. J Neuropathol Exp Neurol 38(5):475–489
Monaco S, Nardelli E, Moretto G, Cavallaro T, Rizzuto N (1988) Cytoskeletal pathology in ataxia-telangiectasia. Clin Neuropathol 7(1):44–46
Volkow ND, Tomasi D, Wang GJ, Studentsova Y, Margus B, Crawford TO (2014) Brain glucose metabolism in adults with ataxia-telangiectasia and their asymptomatic relatives. Brain J Neurol. doi:10.1093/brain/awu092
Baloh RW, Yee RD, Boder E (1978) Eye movements in ataxia-telangiectasia. Neurology 28(11):1099–1104
Lewis RF, Lederman HM, Crawford TO (1999) Ocular motor abnormalities in ataxia telangiectasia. Ann Neurol 46(3):287–295
Reye C (1960) Ataxia-Telangiectasia Report of a Case. AMA J Dis Child 99(2):238–241
Lin DD, Barker PB, Lederman HM, Crawford TO (2014) Cerebral abnormalities in adults with ataxia-telangiectasia. AJNR Am J Neuroradiol 35(1):119–123. doi:10.3174/ajnr.A3646
Navarro C, Martin JJ (1972) Peculiar lesions in Louis-Bar ataxia-telangiectasia. Nucleo-cytoplasmic malformations at the level of ganglionic neuroglia and in a certain number of visceral organs. J Neurol Sci 17(2):219–231
Hernandez D, McConville CM, Stacey M, Woods CG, Brown MM, Shutt P, Rysiecki G, Taylor AM (1993) A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22-23. J Med Genet 30(2):135–140
Klein C, Wenning GK, Quinn NP, Marsden CD (1996) Ataxia without telangiectasia masquerading as benign hereditary chorea. Mov Disord: Off J Mov Disord Soc 11(2):217–220. doi:10.1002/mds.870110217
Delia D, Piane M, Buscemi G, Savio C, Palmeri S, Lulli P, Carlessi L, Fontanella E, Chessa L (2004) MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet 13(18):2155–2163. doi:10.1093/hmg/ddh221
Fernet M, Gribaa M, Salih MA, Seidahmed MZ, Hall J, Koenig M (2005) Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet 14(2):307–318. doi:10.1093/hmg/ddi027
Sekijima Y, Ohara S, Nakagawa S, Tabata K, Yoshida K, Ishigame H, Shimizu Y, Yanagisawa N (1998) Hereditary motor and sensory neuropathy associated with cerebellar atrophy (HMSNCA): clinical and neuropathological features of a Japanese family. J Neurol Sci 158(1):30–37
Barbot C, Coutinho P, Chorao R, Ferreira C, Barros J, Fineza I, Dias K, Monteiro J, Guimaraes A, Mendonca P, do Ceu Moreira M, Sequeiros J (2001) Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch Neurol 58(2):201–205
Shimazaki H, Takiyama Y, Sakoe K, Ikeguchi K, Niijima K, Kaneko J, Namekawa M, Ogawa T, Date H, Tsuji S, Nakano I, Nishizawa M (2002) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia: the aprataxin gene mutations. Neurology 59(4):590–595
Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, Tardieu M, Said G, Habert MO, Demarquay G, Tannier C, Beis JM, Brice A, Koenig M, Durr A (2003) Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain J Neurol 126(Pt 12):2761–2772. doi:10.1093/brain/awg283
Tranchant C, Fleury M, Moreira MC, Koenig M, Warter JM (2003) Phenotypic variability of aprataxin gene mutations. Neurology 60(5):868–870
Habeck M, Zuhlke C, Bentele KH, Unkelbach S, Kress W, Burk K, Schwinger E, Hellenbroich Y (2004) Aprataxin mutations are a rare cause of early onset ataxia in Germany. J Neurol 251(5):591–594. doi:10.1007/s00415-004-0374-7
Amouri R, Moreira MC, Zouari M, El Euch G, Barhoumi C, Kefi M, Belal S, Koenig M, Hentati F (2004) Aprataxin gene mutations in Tunisian families. Neurology 63(5):928–929
Criscuolo C, Mancini P, Menchise V, Sacca F, De Michele G, Banfi S, Filla A (2005) Very late onset in ataxia oculomotor apraxia type I. Ann Neurol 57(5):777. doi:10.1002/ana.20463
D'Arrigo S, Riva D, Bulgheroni S, Chiapparini L, Castellotti B, Gellera C, Pantaleoni C (2008) Ataxia with oculomotor apraxia type 1 (AOA1): clinical and neuropsychological features in 2 new patients and differential diagnosis. J Child Neurol 23(8):895–900. doi:10.1177/0883073808314959
Nemeth AH, Bochukova E, Dunne E, Huson SM, Elston J, Hannan MA, Jackson M, Chapman CJ, Taylor AM (2000) Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxia-telangiectasia-like syndrome) is linked to chromosome 9q34. Am J Hum Genet 67(5):1320–1326. doi:10.1016/S0002-9297(07)62962-0
Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, Goizet C, Moreira MC, Klur S, Yahyaoui M, Agid Y, Koenig M, Stevanin G, Brice A, Durr A (2004) Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain J Neurol 127(Pt 4):759–767. doi:10.1093/brain/awh080
Duquette A, Roddier K, McNabb-Baltar J, Gosselin I, St-Denis A, Dicaire MJ, Loisel L, Labuda D, Marchand L, Mathieu J, Bouchard JP, Brais B (2005) Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol 57(3):408–414. doi:10.1002/ana.20408
Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Sacca F, Grieco GS, Piane M, Barbieri F, De Michele G, Banfi S, Pierelli F, Rizzuto N, Santorelli FM, Gallosti L, Filla A, Casali C (2006) Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 66(8):1207–1210. doi:10.1212/01.wnl.0000208402.10512.4a
Asaka T, Yokoji H, Ito J, Yamaguchi K, Matsushima A (2006) Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology 66(10):1580–1581. doi:10.1212/01.wnl.0000216135.59699.9b
Nicolaou P, Georghiou A, Votsi C, Middleton LT, Zamba-Papanicolaou E, Christodoulou K (2008) A novel c.5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Med Genet 9:28
Tazir M, Ali-Pacha L, M'Zahem A, Delaunoy JP, Fritsch M, Nouioua S, Benhassine T, Assami S, Grid D, Vallat JM, Hamri A, Koenig M (2009) Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci 278(1–2):77–81. doi:10.1016/j.jns.2008.12.004
Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schols L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M'Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M (2009) Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain J Neurol 132(Pt 10):2688–2698. doi:10.1093/brain/awp211
Gomez-Herreros F, Schuurs-Hoeijmakers JH, McCormack M, Greally MT, Rulten S, Romero-Granados R, Counihan TJ, Chaila E, Conroy J, Ennis S, Delanty N, Cortes-Ledesma F, de Brouwer AP, Cavalleri GL, El-Khamisy SF, de Vries BB, Caldecott KW (2014) TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet. doi:10.1038/ng.2929
Al Tassan N, Khalil D, Shinwari J, Al Sharif L, Bavi P, Abduljaleel Z, Abu Dhaim N, Magrashi A, Bobis S, Ahmed H, Alahmed S, Bohlega S (2012) A missense mutation in PIK3R5 gene in a family with ataxia and oculomotor apraxia. Hum Mutat 33(2):351–354. doi:10.1002/humu.21650
Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366(7):636–646. doi:10.1056/NEJMra1006610
Fogel BL (2012) Childhood cerebellar ataxia. J Child Neurol 27(9):1138–1145. doi:10.1177/0883073812448231
Harding AE (1981) Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain J Neurol 104(3):589–620
Koeppen AH (2011) Friedreich's ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 303(1–2):1–12. doi:10.1016/j.jns.2011.01.010
Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 335(16):1169–1175. doi:10.1056/NEJM199610173351601
Harding AE, Matthews S, Jones S, Ellis CJ, Booth IW, Muller DP (1985) Spinocerebellar degeneration associated with a selective defect of vitamin E absorption. N Engl J Med 313(1):32–35. doi:10.1056/NEJM198507043130107
Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, Arai H, Inoue K, Mandel JL, Koenig M (1995) Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet 9(2):141–145. doi:10.1038/ng0295-141
Jayadev S, Bird TD (2013) Hereditary ataxias: overview. Genet Med: Off J Am Coll Med Genet 15(9):673–683. doi:10.1038/gim.2013.28
Verbeek DS, van de Warrenburg BP (2011) Genetics of the dominant ataxias. Semin Neurol 31(5):461–469. doi:10.1055/s-0031-1299785
Matilla-Duenas A, Corral-Juan M, Volpini V, Sanchez I (2012) The spinocerebellar ataxias: clinical aspects and molecular genetics. Adv Exp Med Biol 724:351–374. doi:10.1007/978-1-4614-0653-2_27
Caldecott KW (2008) Single-strand break repair and genetic disease. Nat Rev Genet 9(8):619–631. doi:10.1038/nrg2380
McKinnon PJ (2009) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10(2):100–112. doi:10.1038/nrn2559
Lamarche BJ, Orazio NI, Weitzman MD (2010) The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 584(17):3682–3695. doi:10.1016/j.febslet.2010.07.029
Bensimon A, Aebersold R, Shiloh Y (2011) Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett 585(11):1625–1639. doi:10.1016/j.febslet.2011.05.013
El-Khamisy SF (2011) To live or to die: a matter of processing damaged DNA termini in neurons. EMBO Mol Med 3(2):78–88. doi:10.1002/emmm.201000114
Langerak P, Russell P (2011) Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond Ser B Biol Sci 366(1584):3562–3571. doi:10.1098/rstb.2011.0070
Jeppesen DK, Bohr VA, Stevnsner T (2011) DNA repair deficiency in neurodegeneration. Prog Neurobiol 94(2):166–200. doi:10.1016/j.pneurobio.2011.04.013
Ditch S, Paull TT (2012) The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 37(1):15–22. doi:10.1016/j.tibs.2011.10.002
Hoche F, Seidel K, Theis M, Vlaho S, Schubert R, Zielen S, Kieslich M (2012) Neurodegeneration in ataxia telangiectasia: what is new? What is evident? Neuropediatrics 43(3):119–129. doi:10.1055/s-0032-1313915
McKinnon PJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7:303–321. doi:10.1146/annurev-pathol-011811-132509
Reynolds JJ, Stewart GS (2013) A nervous predisposition to unrepaired DNA double strand breaks. DNA Repair (Amst). doi:10.1016/j.dnarep.2013.04.011
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14(4):197–210. doi:10.1038/nrm3546
McKinnon PJ (2013) Maintaining genome stability in the nervous system. Nat Neurosci 16(11):1523–1529. doi:10.1038/nn.3537
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316(5828):1160–1166. doi:10.1126/science.1140321
Takahashi T, Tada M, Igarashi S, Koyama A, Date H, Yokoseki A, Shiga A, Yoshida Y, Tsuji S, Nishizawa M, Onodera O (2007) Aprataxin, causative gene product for EAOH/AOA1, repairs DNA single-strand breaks with damaged 3′-phosphate and 3′-phosphoglycolate ends. Nucleic Acids Res 35(11):3797–3809. doi:10.1093/nar/gkm158
Pouliot JJ, Yao KC, Robertson CA, Nash HA (1999) Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 286(5439):552–555
Interthal H, Chen HJ, Champoux JJ (2005) Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem 280(43):36518–36528. doi:10.1074/jbc.M508898200
Lebedeva NA, Rechkunova NI, Lavrik OI (2011) AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett 585(4):683–686. doi:10.1016/j.febslet.2011.01.032
Lebedeva NA, Rechkunova NI, El-Khamisy SF, Lavrik OI (2012) Tyrosyl-DNA phosphodiesterase 1 initiates repair of apurinic/apyrimidinic sites. Biochimie 94(8):1749–1753. doi:10.1016/j.biochi.2012.04.004
Lebedeva NA, Rechkunova NI, Ishchenko AA, Saparbaev M, Lavrik OI (2013) The mechanism of human tyrosyl-DNA phosphodiesterase 1 in the cleavage of AP site and its synthetic analogs. DNA Repair (Amst). doi:10.1016/j.dnarep.2013.09.008
Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW (2001) XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104(1):107–117
Plo I, Liao ZY, Barceló JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y (2003) Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2(10):1087–1100
Koch CA, Agyei R, Galicia S, Metalnikov P, O'Donnell P, Starostine A, Weinfeld M, Durocher D (2004) Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J 23(19):3874–3885. doi:10.1038/sj.emboj.7600375
Reynolds JJ, Walker AK, Gilmore EC, Walsh CA, Caldecott KW (2012) Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic Acids Res 40(14):6608–6619. doi:10.1093/nar/gks318
Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, Gatei M, Criscuolo C, Filla A, Chessa L, Fusser M, Epe B, Gueven N, Lavin MF (2007) Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 177(6):969–979. doi:10.1083/jcb.200701042
Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, Lavin MF (2009) Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet 18(18):3384–3396. doi:10.1093/hmg/ddp278
De Amicis A, Piane M, Ferrari F, Fanciulli M, Delia D, Chessa L (2011) Role of senataxin in DNA damage and telomeric stability. DNA Repair (Amst) 10(2):199–209. doi:10.1016/j.dnarep.2010.10.012
Skourti-Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42(6):794–805. doi:10.1016/j.molcel.2011.04.026
Yuce O, West SC (2013) Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 33(2):406–417. doi:10.1128/MCB.01195-12
Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, Zhao J, Russell HR, Petrini JH, Nitiss JL, McKinnon PJ (2014) Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat Neurosci 17(6):813–821. doi:10.1038/nn.3715
Elia M (1991) Organ and tissue contribution to metabolic rate. In: John M, Kinney HNT (eds) Energy Metabolism: Tissue Determinants and Cellular Corollaries. Raven Press, Ltd., New York, pp 61–79
Dolle ME, Snyder WK, Dunson DB, Vijg J (2002) Mutational fingerprints of aging. Nucleic Acids Res 30(2):545–549
Maslov AY, Ganapathi S, Westerhof M, Quispe-Tintaya W, White RR, Van Houten B, Reiling E, Dolle ME, van Steeg H, Hasty P, Hoeijmakers JH, Vijg J (2013) DNA damage in normally and prematurely aged mice. Aging Cell 12(3):467–477. doi:10.1111/acel.12071
Acevedo-Torres K, Berrios L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, Torres-Ramos CA, Ayala-Torres S (2009) Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease. DNA Repair (Amst) 8(1):126–136. doi:10.1016/j.dnarep.2008.09.004
Canugovi C, Misiak M, Ferrarelli LK, Croteau DL, Bohr VA (2013) The role of DNA repair in brain related disease pathology. DNA Repair (Amst) 12(8):578–587. doi:10.1016/j.dnarep.2013.04.010
Chen H, Chan DC (2009) Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 18(R2):R169–R176. doi:10.1093/hmg/ddp326
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337(6098):1062–1065. doi:10.1126/science.1219855
Stern N, Hochman A, Zemach N, Weizman N, Hammel I, Shiloh Y, Rotman G, Barzilai A (2002) Accumulation of DNA damage and reduced levels of nicotine adenine dinucleotide in the brains of Atm-deficient mice. J Biol Chem 277(1):602–608. doi:10.1074/jbc.M106798200
Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS (2007) Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 117(9):2723–2734. doi:10.1172/JCI31604
Ambrose M, Goldstine JV, Gatti RA (2007) Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet 16(18):2154–2164. doi:10.1093/hmg/ddm166
Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB (2012) Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119(6):1490–1500. doi:10.1182/blood-2011-08-373639
Stoykova AS, Dudov KP, Dabeva MD, Hadjiolov AA (1983) Different rates of synthesis and turnover of ribosomal RNA in rat brain and liver. J Neurochem 41(4):942–949
Stoykova AS, Dabeva MD, Dimova RN, Hadjiolov AA (1985) Ribosome biogenesis and nucleolar ultrastructure in neuronal and oligodendroglial rat brain cells. J Neurochem 45(6):1667–1676
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gilmore, E.C. DNA repair abnormalities leading to ataxia: shared neurological phenotypes and risk factors. Neurogenetics 15, 217–228 (2014). https://doi.org/10.1007/s10048-014-0415-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-014-0415-z