
Abstract
Episodic ataxia is an autosomal dominant ion channel disorder characterized by paroxysmal attacks of incoordination. Episodic ataxia type 2 (EA2) is caused by mutations in CACNA1A. EA2 mutations are mostly nonsense and sometimes missense mutations. However, in some typical EA2 families, CACNA1A sequencing does not detect any point mutation. Herein, we have designed a quantitative multiplex polymerase chain reaction of short fluorescent fragment test to screen the 50 exons of CACNA1A and investigated 27 probands referred for molecular diagnosis of EA2 who did not show any point mutation in CACNA1A. We have identified four different exonic deletions in four patients with a typical EA2 phenotype. These results establish the need to complete sequencing analysis by a screening for deletions to ensure an accurate molecular diagnosis of EA2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Episodic ataxia type 2 (EA2 [MIM 108500]) is an autosomal dominant neurological disorder, characterized by early onset recurrent attacks of incoordination lasting hours to days with interictal eye movement abnormalities. These attacks are often triggered by fatigue or emotional stress and are most often responsive to acetazolamide [1]. EA2 is caused by mutations in CACNA1A [2]. CACNA1A [MIM 601011] encodes the CaV2.1 subunit, which is the pore-forming and voltage-sensing subunit of a neuronal P/Q type calcium channel expressed throughout the CNS but most abundant in the cerebellum [3]. EA2 has been shown to be associated most often with nonsense and sometimes with missense mutations within CACNA1A. More than 40 different point mutations have been published [1, 4, 5]. However, in some typical EA2 families, CACNA1A screening does not detect any point mutation [4–7]. The loss of function nature of nonsense mutations suggest that deletions may be involved in this disease. We have recently described the first EA2 family with a large deletion in CACNA1A gene [8]. Herein, we report four different novel deletions of CACNA1A in 4 EA2 probands.
Materials and methods
Study subjects
The study included 27 patients referred for molecular diagnosis of EA2. They all presented several bouts of ataxia or vertigo, consistent with the diagnosis of paroxysmal ataxia. Blood samples had been collected after obtaining written informed consent. Sequencing of the 50 exons of CACNA1A had been previously performed and did not detect any point mutation.
QMPSF conditions
The quantitative multiplex polymerase chain reaction (PCR) of short fluorescent fragments (QMPSF) method is described in detail elsewhere [9]. Oligonucleotide primer pairs for amplification of short fluorescent fragments corresponding to CACNA1A exons were designed using the Primer Premier 5 Software (Primer Biosoft International, Palo Alto, CA, USA). Six multiplex PCRs were set up to check the copy number of the 50 exons of CACNA1A. All forward primers were 5′-labeled with the 6-FAM fluorochrome. Each multiplex PCR set contained a control primer set that amplified a short sequence of HMBS (hydroxymethylbilane synthase gene), not involved in EA2. (Primer sequences are available on request.) Amplicon sizes ranged between 120 and 250 bp. All DNA samples were re-extracted using the QIAamp DNA blood mini kit (Qiagen Hilden, Germany). Reactions were done as described previously [10].
PCR products were separated by capillary electrophoresis on an ABI 3130 Genetic Analyser (Applied Biosystems Foster City, USA). Quantification of the area of peaks corresponding to the tested exons and to the internal HMBS control was determined using GeneMapper analysis software version 4.0 (Applied Biosystems). The copy number of each tested exon was expressed as the following ratio: (area of the peak corresponding to a tested exon for the patient/area of the peak corresponding to HMBS for the patient)/(area of the peak corresponding to a tested exon for the control DNA/area of the peak corresponding to HMBS for the control DNA). Calculation of ratios was done by the software. A ratio close to 1 is obtained when two copies of the exon are present and a ratio close to 0.5 when only one copy of the tested exon is present (hemizygous DNA).
Long-range PCR and sequencing
Long-range PCR amplifications was performed with the TripleMaster® PCR System (Eppendorf AG—Hamburg, Germany) according to the manufacturer recommendations. Sequencing was performed using standard protocols, on an ABI 3130 DNA analyser (Applied Biosystems).
Results
The 27 DNA samples underwent QMPSF analysis. Four patients presented a ratio close to 0.5 for one or several exons, consistent with an exonic deletion. Ratios were close to 1 for the 50 exons of the 23 remaining patients.
Clinical data of the four probands with a deletion
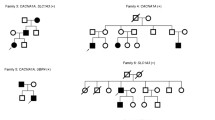
Pedigrees and clinical data of the four deleted probands and their available relatives are summarized in Fig. 1 and Table 1. Briefly, all four probands showed typical episodic ataxia episodes. Age of onset ranged between 2 and 19 years old; acetazolamide was effective in all of them. Two of the probands (1 and 3) had at least one relative affected with the same symptoms. One (proband 2) was a sporadic case. His parents were aged over 60 and never had any clinical manifestation; their DNA was not available. The last one (proband 4) did not had relatives with typical EA2, but his mother had presented atypical clinical symptoms, including bilateral hypoacusia, rotatory vertigo episodes, and tinnitus. Clinical examination showed a mild nystagmus. She had epilepsy and learning disabilities during childhood.

Pedigrees of the four probands with a CACNA1A deletion: circle females, squares males, arrows probands, filled symbols episodic ataxia affected, empty symbols clinically healthy, striped neurological symptoms of unknown significance, question marks no information available, crossed deceased, letter T DNA available, +/del heterozygous deletion
Molecular data
Ratios obtained by QMPSF analysis for patient 1 were close to 0.5 for exons 38, 39, and 40 and were close to 1 for the other exons. Long-range PCR was performed to determine the size and breakpoints of the deletion and sequence analysis of a fragment spanning the deleted region revealed that the deletion was 12,143 bp in size. The deletion IVS37_IVS40 removes 1,151 bp of intron 37 adjacent to exon 38, all of exon 38 to exon 40, and 452 bp of intron 40 adjacent to exon 40 (Fig. 2a). The same deletion was detected in his mother. The open-reading frame should be maintained after the deletion since the cumulated size of the deleted exons is 315 bp (105 codons).

Breakpoints of the deletions: a the deletion identified in patient 1 begins after the 5,628th nucleotide of IVS37, removes 1,151 bp of IVS37 adjacent to exon 38, all of exon 38 to exon 40, and 452 bp of IVS40 adjacent to exon 40. b Deletion identified in patient 2 begins after the 21,243rd nucleotide of IVS6, removes 2,453 bp of IVS6 adjacent to exon 7, all exons 7 to 10, and 3,157 bp of IVS10 adjacent to exon 10. c Deletion identified in patient 3 begins after the 448th nucleotide of IVS29, removes 1,545 bp of IVS29 adjacent to exon 30, exon 30, and 2,385 bp of IVS30 adjacent to exon 30. DNA repair was accompanied by an insertion of 22 bp where the DNA breakpoints rejoined. d Deletion identified in patient 4 begins after the 5,517th nucleotide of IVS5, removes 2,649 bp of IVS5 adjacent to exon 6, exon 6 and 14,740 bp of IVS6 adjacent to exon 6
Ratios obtained by QMPSF analysis for patient 2 were close to 0.5 for exons 7, 8, 9, and 10 and were close to 1 for the other exons. Long-range PCR and sequence analysis revealed that the deletion was 11,276 bp in size. The deletion, IVS6_IVS10 removes 2,453 bp of intron 6 adjacent to exon 7, all of exon 7 to exon 10, and 3,157 bp of intron 10 adjacent to exon 10 (Fig. 2b). The deletion should lead to a frameshift and a premature stop codon.
Ratios obtained by QMPSF analysis for patient 3 were close to 0.5 for exons 30 and were close to 1 for all other exons. Long-range PCR and sequencing revealed that the deletion size was 4,041 bp. The deletion, IVS29_IVS30, removes 1,545 bp of intron 29 adjacent to exon 30, exon 30, and 2,385 bp of intron 30 adjacent to exon 30. DNA repair was accompanied by an insertion of 22 bp where the DNA breakpoints rejoined (Fig. 2c). As patient 1, the open-reading frame is conserved after the deletion, the size of exon 30 being 111 bp (37 codons).
Ratios obtained by QMPSF analysis for patient 4 were close to 0.5 for exons 6 and close to 1 for the other exons. Long-range PCR and sequencing revealed that the deletion size was 17,583 bp. The deletion, IVS5_IVS6, removes 2,649 bp of intron 5 adjacent to exon 6, exon 6, and 14,740 bp of intron 6 adjacent to exon 6 (Fig. 2c). The deletion should lead to a frame shift and a premature stop codon. The same deletion was present in his mother. These four deletions were absent in 102 control individuals.
To determine if these deletions arose from a common mechanism, analysis of the sequence around both breakpoints of each deletion was performed to search for homologies and repeated domains with the RepeatMasker Software (Institute for System Biology, Seattle, WA, USA; Fig. 3). The proximal breakpoint of patient 1 was located within a FLAM-C sequence (chr19: 13197804-13197678) and the distal breakpoint within an Alu repeat (chr19: 13785799-13185487). The proximal breakpoint for patient 2 was not located in a repeated sequence, but the distal breakpoint was located within an Alu sequence (ch 19: 13298994-13298710). Patient 3′ proximal breakpoint was located within an Alu sequence (chr19: 13226474-13226164), and the distal breakpoint was within an Alu repeat (chr19: 13222509-132222215). The insertion of 22 bp at the breakpoint corresponds to an Alu sequence. Patient 4 had a proximal breakpoint and a distal breakpoint within two Alu sequences (respectively, chr19: 13334010-13334316 and chr19: 13316589-13316872). Homology between the two sequences on both sides of the deletion (on the 30 last bases before the recombination site) was 90% for patients 1 and 4 and lower for patients 2 and 3 (respectively, 26% and 50%).

Sequence alignment at the breakpoints. Each mutated sequence is aligned with the breakpoint corresponding sequences: introns 37 and 40 for patient 1, introns 6 and 10 for patient 2, introns 29 and 30 for patient 3, and introns 5 and 6 for patient 4. Boxed identical nucleotides between the two intronic sequences at the breakpoints, underlined the 22 bp insertion at the patient 3′ breakpoint. Alu sequences are noted in blue
Discussion
We performed a QMPSF analysis of CACNA1A in 27 probands referred for EA2 genetic testing and in whom no point mutation had been identified by sequencing. This analysis allowed the detection of four different rearrangements in four probands, two deletions of a single exon, one deletion of three exons, and one of four exons. The four probands had a typical EA2 phenotype. Three cases were familial (patients 1, 3, and 4), and one case was clinically sporadic (patient 2).
Two of the deletions are predicted to lead to a frameshift and a premature stop codon (patients 2 and 4). The consequence of these two mutations is identical to those of previously reported premature stop codons observed in EA2 families, the deleted alleles should lead either to a shorter protein or an absence of protein due to mRNA decay. The other two are predicted to be in frame deletions. The consequences of these deletions cannot be firmly established because neither the mRNA nor the protein encoded by this gene is available for study. Two hypotheses may at least be raised. Although in frame, these deletions may lead to an absence of the mutated protein at the cell membrane through protein instability or misrouting, the consequences would then be similar to classical loss of function mutations. Alternatively, these two in-frame deletions might lead to shorter Cav2.1 proteins expressed at the cell surface. Exon 30 (deleted in patient 3) encodes the major part of segment IV-S2 and the loop between IV-S1 and IV-S2. The absence of this exon will alter the structure of the fourth domain and should affect the correct assembly of the protein in the membrane. The other in-frame deletion (deletion of exons 38–39–40 found in family 1) removes 105 amino acids in the proximal half of the cytoplasmic C-terminal tail of the protein (p.Arg1877_Gln1981). This region contains the IQ domain implicated in Cav2.1 regulation that is essential for channel function, in particular Ca2+-dependant facilitation and inactivation [11, 12].
Deletions seem to be highly heterogeneous both in their location and size (4.0 to 17.5 kb); however, the breakpoints are mostly located in Alu elements. Alu elements are the most abundant class of interspersed repeat sequences, covering 5–10% of the genome and occurring on average once every 4 kb [13]. Alu repeats are known to favor recombination events, either by homologous or nonhomologous recombination. Alu sequences have been implicated in many diseases, including cancers [14, 15]. In CACNA1A, they are over-represented as compared to the average in the human genome, corresponding to 29.2% of the gene sequence (327 Alu sequences). So, in EA2, recombination events in CACNA1A might be favored by the high number of Alu elements.
To summarize, we have identified a causative deletion within CACNA1A in four of 27 (14%) patients referred for molecular testing and in whom no point mutation was identified. These results emphasize the need to complete sequencing analysis by screening for deletions to increase the sensitivity of molecular test. However, we did not detect a point mutation nor a deletion in the remaining 23 patients, suggesting the existence of mutations located in the intronic or regulatory regions of the gene, or genetic heterogeneity since other types of episodic ataxia had been reported that can be clinically similar to type 2 [16].
References
Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW (2007) Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 130:2484–2493
Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM et al (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87:543–552
Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P et al (1991) Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature 350:398–402
Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A et al (1999) High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 52:1816–1821
Jen J, Kim GW, Baloh RW (2004) Clinical spectrum of episodic ataxia type 2. Neurology 62:17–22
Mantuano E, Veneziano L, Spadaro M, Giunti P, Guida S, Leggio MG et al (2004) Clusters of non-truncating mutations of P/Q type Ca2+ channel subunit Ca(v)2.1 causing episodic ataxia 2. J Med Genet 41:e82
Eunson LH, Graves TD, Hanna MG (2005) New calcium channel mutations predict aberrant RNA splicing in episodic ataxia. Neurology 65:308–310
Riant F, Mourtada R, Saugier-Veber P, Tournier-Lasserve E (2008) Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol 65:817–820
Casilli F, Di Rocco ZC, Gad S, Tournier I, Stoppa-Lyonnet D, Frebourg T et al (2002) Rapid detection of novel BRCA1 rearrangements in high-risk breast-ovarian cancer families using multiplex PCR of short fluorescent fragments. Hum Mutat 20:218–226
Saugier-Veber P, Goldenberg A, Drouin-Garraud V, de La Rochebrochard C, Layet V, Drouot N et al (2006) Simple detection of genomic microdeletions and microduplications using QMPSF in patients with idiopathic mental retardation. Eur J Hum Genet 14:1009–1017
DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411:484–489
Dunlap K (2007) Calcium channels are models of self-control. J Gen Physiol 129:379–383
Kolomietz E, Meyn MS, Pandita A, Squire JA (2002) The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 35:97–112
Deininger PL, Batzer MA (1999) Alu repeats and human disease. Mol Genet Metab 67:183–193
van Zelm MC, Geertsema C, Nieuwenhuis N, de Ridder D, Conley ME, Schiff C et al (2008) Gross deletions involving IGHM, BTK, or Artemis: a model for genomic lesions mediated by transposable elements. Am J Hum Genet 82:320–332
Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW (2007) A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol 64:749–752
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Riant, F., Lescoat, C., Vahedi, K. et al. Identification of CACNA1A large deletions in four patients with episodic ataxia. Neurogenetics 11, 101–106 (2010). https://doi.org/10.1007/s10048-009-0208-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-009-0208-y