
Abstract
Autosomal dominant episodic ataxia type 2 (EA2) is caused by variants in CACNA1A. We examined a 20-year-old male with EA symptoms from a Japanese family with hereditary EA. Cerebellar atrophy was not evident, but single photon emission computed tomography showed cerebellar hypoperfusion. We identified a novel nonsynonymous variant in CACNA1A, NM_001127222.2:c.1805T>G (p.Leu602Arg), which is predicted to be functionally deleterious; therefore, this variant is likely responsible for EA2 in this pedigree.
Similar content being viewed by others
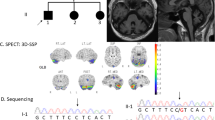
Hereditary episodic ataxia (EA) is a heterogeneous group of movement disorders characterized by recurrent spells of truncal ataxia and incoordination1. EA type 2 (EA2, MIM: 108500) is an autosomal dominant hereditary EA caused by heterozygous variants in the calcium voltage-gated channel subunit alpha-1A gene (CACNA1A, MIM: 601011). Here, we studied a patient with hereditary EA originating from Shikoku Island, Japan (Fig. 1A). The proband (Patient III-1) was a 20-year-old male who had experienced brief episodes (<30 min) of ataxia since childhood that were precipitated by actions such as running or riding a bicycle. The frequency of the episodes was once a week at most. Witnesses said that his eyes were bloodshot while he was experiencing EA. Interictal neurological examination showed no abnormalities. Brain magnetic resonance imaging (MRI) did not show obvious cerebellar atrophy (Fig. 1B). Interictal brain single photon emission computed tomography (SPECT) using N-isopropyl-p-(iodine-123)-iodoamphetamine (123I-IMP) with three-dimensional stereotactic surface projections showed hypoperfusion in the cerebellum, brainstem, and lateral occipital lobe (Fig. 1C). His symptoms were initially improved with acetazolamide (125 mg/day), but the effect did not last. According to the proband, his mother (II-3) and maternal grandfather (I-1) experienced the same ataxic symptoms until they were 20 years old. His 16-year-old younger brother (III-2) and 9-year-old sister (III-3) also exhibit the same ataxic symptoms.

A Pedigree of the EA2 family. Squares: males, circles: females, solid symbols: affected individuals, open symbols: unaffected individuals. An arrow indicates the proband. B Brain magnetic resonance imaging (MRI) did not show obvious cerebellar atrophy. C Single photon emission computed tomography (SPECT) using N-isopropyl-p-(iodine-123)-iodoamphetamine (123I-IMP) with three-dimensional stereotactic surface projections showed hypoperfusion in the cerebellum, brainstem and lateral occipital lobe. R right, L left, RT. LAT right lateral, LT. LAT left lateral, SUP superior, INF inferior, ANT anterior, POST posterior, RT. MED right medial, LT. MED left medial, Z Z score.
We first confirmed the absence of repeat expansion in genes known to be responsible for spinocerebellar ataxia (SCA) 1–3, 6–8, 10, 12, 17, and 36 and dentatorubral-pallidoluysian atrophy. We also confirmed the absence of pathogenic variants in SCA31. We then performed exome sequencing of the proband using SureSelect Human All Exon v6 (Agilent Technologies, Santa Clara, CA, USA) on the Illumina NovaSeq 6000 platform (Illumina, Inc., San Diego, CA, USA). We achieved a sequencing depth of 168× and identified 24,655 variants in the proband. Since the proband was diagnosed with EA, we selected 21 variants located in eight genes known to be associated with EA (Supplementary Table 1). By excluding variants already registered in public databases [the 1000 Genomes Project (http://www.1000genomes.org), ExAC (http://exac.broadinstitute.org/), and gnomAD (https://gnomad.broadinstitute.org/)], we identified a novel nonsynonymous variant located in exon 14 of the calcium voltage-gated channel subunit alpha-1A gene (CACNA1A), NM_001127222.2:c.1805T>G (p.Leu602Arg) (Fig. 2). The variant was predicted to be “probably damaging” by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), “deleterious” by SIFT (https://sift.bii.a-star.edu.sg/) and “disease-causing” by Mutation Taster (https://www.mutationtaster.org) with a CADD score of 29.4. Given the patient’s symptoms, genes known to be associated with dystonia, episodic kinesigenic dyskinesia and SCAs were also examined (Supplementary Table 1). However, no pathological variants were detected in any genes associated with these three conditions.

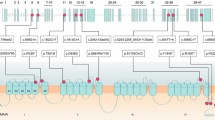
A Electropherogram of the CACNA1A NM_001127222.2:c.1805T>G (p.Leu602Arg) variant region in the proband (III-1) and an unaffected control. The location of the variant is indicated by a red arrow. B Genomic structure of the CACNA1A gene. The ion transport domain (IT), voltage-dependent L-type calcium channel, IQ-associated domain (GPTH) and voltage-gated calcium channel IQ domain (Ca_chan_IQ) are shown according to the NCBI Conserved Domain Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?seqinput=NP_001120693.1). Red arrows indicate the location of the variant.
We validated the CACNA1A variant in the patient by Sanger sequencing (forward primer, 5′-GGGAAAGTGAGCCTCGTGT-3′ and reverse primer 5′-GGAGTTGGAATTCCTGTGAAG-3′). We confirmed that the patient was heterozygous for the variant, consistent with the autosomal dominant mode of disease inheritance (Fig. 2A). We also examined the CAG repeat length in CACNA1A and confirmed that the proband was homozygous for an 11-repeat allele that is nonpathogenic. According to the ACMG/AMP/CAP guidelines, the p.Leu602Arg variant is classified as “likely pathogenic”, meeting the PM1, PM2, PP3 and PP4 criteria2. The CACNA1A variant data have been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/1809801/).
We described here a Japanese EA2 patient carrying a novel nonsynonymous heterozygous variant [CACNA1A, NM_001127222.2:c.1805T > G (p.Leu602Arg)]. The patient exhibited a typical EA2 phenotype1,3. The neuroradiological features of our patient included hypoperfusion of the cerebellum on brain SPECT despite no marked cerebellar atrophy on brain MRI. Brain SPECT is a functional neuroimaging technique for evaluating cerebrovascular disorders, neurodegenerative diseases, and epilepsy and may be able to detect lesions that do not produce abnormal findings on MRI4,5. As in the present case, brain MRI revealed no cerebellar atrophy in other EA2 cases6,7. To our knowledge, this is the first report of brain SPECT in a patient with EA2 caused by a heterozygous point mutation in the CACNA1A gene, although a brain SPECT study has been reported for a patient with familial hemiplegic migraine 1 (FHM1, MIM: 141500) carrying a heterozygous point mutation in the CACNA1A gene8. Additionally, several brain SPECT studies in patients with SCA6 (MIM: 183086) caused by a CAG trinucleotide repeat expansion in the CACNA1A gene have been reported9,10,11. All reported patients with CACNA1A variations showed atrophy and hypoperfusion localized in the cerebellum8,9,10,11. There are no previous reports of brain SPECT in patients with EA2; therefore, it is currently unclear whether reduced perfusion of the brainstem is common. It is important to collect brain SPECT data from more EA2 patients. The nonsynonymous variant is located within one of the ion-transport domains that is essential for the channel function of the CACNA1A protein (Fig. 2B). Although the nonsynonymous variant is predicted to be highly pathogenic by multiple prediction tools, including “probably damaging” by PolyPhen-2 and “disease-causing” by Mutation Taster, we were unable to perform segregation analysis of the variant because additional family members were not available.
We conclude that the novel CACNA1A variant NM_001127222.2:c.1805T>G (p.Leu602Arg) is highly likely to be responsible for EA2 in the current pedigree. Clinically, when patients complain of recurrent ataxic episodes, it is important to look for cerebellar hypoperfusion by brain SPECT even when brain MRI does not show cerebellar atrophy.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3357.
References
Choi, K. D. & Choi, J. H. Episodic ataxias: clinical and genetic features. J. Mov. Disord. 9, 129–135 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Amadori, E. et al. Genetic paroxysmal neurological disorders featuring episodic ataxia and epilepsy. Eur. J. Med. Genet. 65, 104450 (2022).
Holman, B. L. & Devous, M. D. Functional brain SPECT: the emergence of a powerful clinical method. J. Nucl. Med. 33, 1888–1904 (1992).
Nagamitsu, S. et al. Decreased cerebellar blood flow in postinfectious acute cerebellar ataxia. J. Neurol. Neurosurg. Psychiatry. 67, 109–112 (1999).
Angelini, C. et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur. J. Med. Genet. 62, 103530 (2019).
Verriello, L. et al. Epilepsy and episodic ataxia type 2: family study and review of the literature. J. Neurol. 268, 4296–4302 (2021).
Takahashi, T. et al. Japanese cases of familial hemiplegic migraine with cerebellar ataxia carrying a T666M mutation in the CACNA1A gene. J. Neurol. Neurosurg. Psychiatry. 72, 676–677 (2002).
Nanri, K. et al. Classification of cerebellar atrophy using voxel-based morphometry and SPECT with an easy Z-score imaging system. Inter. Med. 49, 535–541 (2010).
Honjo, K. et al. Quantitative assessment of cerebral blood flow in genetically confirmed spinocerebellar ataxia type 6. Arch. Neurol. 61, 933–937 (2004).
Koh, S. H. et al. Spinocerebellar ataxia type 6 and episodic ataxia type 2 in a Korean family. Korean Med. Sci. 16, 809–813 (2001).
Acknowledgements
This work was supported by Grants-in-Aid from MEXT, Japan (#23K06853) and the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. We thank Jeremy Allen, PhD, from Edanz (https://jp.edanz.com/ac) for English language correction of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miura, S., Watanabe, E., Senzaki, K. et al. Episodic ataxia type 2 with a novel missense variant (Leu602Arg) in CACNA1A. Hum Genome Var 11, 3 (2024). https://doi.org/10.1038/s41439-023-00261-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00261-w
- Springer Nature Limited