
Abstract
A solid-state reaction process with poly(vinyl alcohol) as the carbon source is developed to synthesize LiFePO4-based active powders with or without modification assistance of a small amount of Li3V2(PO4)3. The samples are analyzed by X-ray diffraction, scanning/transmission electron microscopy, and Raman spectroscopy. It is found that, in addition to the minor effect of a lattice doping in LiFePO4 by substituting a tiny fraction of Fe2+ ions with V3+ ions, the change in the form of carbon coating on the surface of LiFePO4 plays a more important role to improve the electrochemical properties. The carbon changes partially from sp3 to sp2 hybridization and thus causes the significant rise in electronic conductivity in the Li3V2(PO4)3-modified LiFePO4 samples. Compared with the carbon-coated baseline LiFePO4, the composite material 0.9LiFePO4·0.1Li3V2(PO4)3 shows totally different carbon morphology and much better electrochemical properties. It delivers specific capacities of 143.6 mAh g−1 at 10 C rate and 119.2 mAh g−1 at 20 C rate, respectively. Even at the low temperature of −20 °C, it delivers a specific capacity of 118.4 mAh g−1 at 0.2 C.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recently, olivine-structure phosphates LiMPO4 (M=Fe, Mn, Co, and Ni) are attractive cathode materials for their stable structure, very flat charge-discharge plateaus, and relatively high capacities [1,2,3,4,5,6]. Among these phosphates, the redox potentials of Mn2+/Mn3+, Co2+/Co3+, and Ni2+/Ni3+ are higher than Fe2+/Fe3+, so LiMPO4 (M=Mn, Co, and Ni) show higher energy density than LiFePO4. Nevertheless, LiCoPO4 and LiNiPO4 have too high operating potentials (4.8 and 5.1 V) that may decompose the electrolyte, while LiMnPO4 suffers from a Jahn-Teller effect in the charge-discharge process that results in capacity fading [7]. Hence, LiFePO4 has been the most popular olivine cathode material for practical applications. On the other hand, because LiMPO4 (M=Fe, Mn, Co, and Ni) materials all suffer from very low intrinsic electronic conductivities and Li-ion diffusivities [8, 9], we have to resort to nano-sizing particles [10, 11], carbon coating [12, 13], and partial cation substitution [14,15,16] to overcome the above drawbacks.
As a desirable carbon coating material on LiFePO4 (LFP) particles, graphene is usually mixed mechanically in electrodes [17], but such a mechanical mixing needs a relatively high graphene content. In a rare case, graphene coating can be also formed in situ with the help of catalytic effect of elemental Fe [18], but Fe is known to be harmful to the electrode. Hence, finding an effective approach to synthesize an in situ graphene-decorated LiFePO4 materials is necessary.
Recently, the synergetic effect of two active components in one electrode has attracted much attention [19, 20]. For example, xLiFePO4·yLi3V2(PO4)3 composites have been prepared by various approaches such as solid-state reaction [21], spray drying [22], sol-gel process [23], and template method [24]. Wang et al. have found that LiFePO4-based cathode with a composition 8LiFePO4·Li3V2(PO4)3 exhibits the highest discharge capacity due to the fact that Li3V2(PO4)3 (LVP) is more ionically conductive than LiFePO4 for its open Nasicon-type structure [25]. They have also attributed the improvement to the possible mutual doping effect of V3+ in LiFePO4 and Fe2+ in Li3V2(PO4)3 within the xLiFePO4·yLi3V2(PO4)3 composites [19, 21, 26]. However, except for the lattice doping effect, a possibly more important reason for the property improvement is rarely noticed, i.e., the change in the form of carbon coating with or without incorporating a small amount of Li3V2(PO4)3 in LiFePO4 during the synthesis. In fact, this is the main goal and the key finding of this study. We attempt to prove it as a primary cause for the improvement. By means of structural analyses, we can successfully identify the change in the form of carbon coating. Also, as a result of this investigation, we have synthesized (1-x)LiFePO4·xLi3V2(PO4)3 composites with both excellent high rate and low-temperature performances compared with a baseline LiFePO4.
Experimental
Synthesis of samples
An intermediate powder for LiFePO4 was synthesized through a solid-state reaction route. Briefly, stoichiometric amounts of CH3COOLi·2H2O (A.R.), FeC2O4·2H2O (A.R.), and NH4H2PO4 (A.R.) with a molar ratio Li:Fe:P = 1:1:1 were dispersed in acetone and ball-milled for 6 h. Subsequently, the slurry was dried and calcined at 500 °C for 11 h under H2 (5%)/Ar to obtain the intermediate powder for LiFePO4.
Meanwhile, another intermediate powder for Li3V2(PO4)3 was synthesized with a similar procedure. Stoichiometric amounts of CH3COOLi·2H2O (A.R.), NH4VO3 (A.R.), and NH4H2PO4 (A.R.) with a molar ratio Li:V:P = 3:2:3 was dispersed in acetone. Citric acid (15 wt%) was also added to act as a reductant. The slurry was ball-milled for 6 h before drying and then calcined at 400 °C for 6 h under H2 (5%)/Ar to obtain the intermediate powder for Li3V2(PO4)3.
The carbon-coated LiFePO4-based powders with compositions (1-x)LiFePO4·xLi3V2(PO4)3 (x = 0, 0.02, 0.05, and 0.1) were synthesized through a two-step solid-state reaction process. Specifically, the above obtained intermediate powders for LiFePO4 and Li3V2(PO4)3 were mixed in a mortar with the mass ratios of 100:0, 98:2, 95:5, and 90:10, respectively. Then polyvinyl alcohol (PVA, with a polymerization degree of 2400) (8 wt%) was added in these mixtures and mixed in the mortar. Finally, these powders were calcined in a tube furnace at 750 °C for 12 h under H2 (5%)/Ar to obtain four samples: LFP@C (sample A), 0.98LFP·0.02LVP@G (sample B), 0.95LFP·0.05LVP@G (sample C), and 0.9LFP·0.1LVP@G (sample D), respectively. The heating rate was 3 °C min−1.
Morphology and structure characterization
The crystallographic information of samples A–D was analyzed by X-ray diffraction (XRD, Rigaku) with Cu Kα radiation over the 2θ angles from 10° to 80°. They were also observed with a scanning electron microscope (SEM, JSM-6390 LA, JEOL) and a (high-resolution) transmission electron microscope (TEM, HRTEM, JEM-2010). The elemental carbon contents in these samples were determined with an infrared carbon-sulfur analyzer (CS-8800C, Jinbo). Raman spectroscopy analysis was performed with a Renishaw inVia Raman microscope (532-nm diode laser excitation). Moreover, X-ray photoelectron spectroscopy (XPS) was measured with ESCALAB 250 (Thermo-VG Scientific).
Electrochemical measurements
The electrochemical properties of these four samples were evaluated using coin-type half-cells (CR2032 size) assembled in an argon-filled glove box (MBraun Labmaster 130). The electrolyte was 1 M LiPF6 in ethylene carbonate and dimethyl carbonate (1:1, v/v), while a micro-porous film Celgard 2400 polypropylene membrane was the separator. The working electrodes were prepared by dispersing active materials (80 wt%), acetylene black (10 wt%), and poly(vinylidene difluoride) (10 wt%) in N-methyl-2-pyrrolidone to obtain homogeneous slurries, which were uniformly coated on an aluminum foil. The cells were evaluated on an electrochemical work station (Chenhua Co., CHI 660C) for cyclic voltammetry (CV) (voltage range of 2.2–4.3 V) and electrochemical impedance spectroscopy (EIS) (frequency 10 mHz to 100 kHz). They were also cycled galvanostatically on a battery cycler (Neware Co., Shenzhen, China) in a voltage range of 2.2–4.3 V at room temperature (25 ± 2 °C) and low temperature (−20 °C), respectively.
Results and discussion
Structures of the (1-x)LiFePO4·xLi3V2(PO4)3 (x = 0, 0.02, 0.05, and 0.1) powders
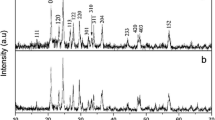
Figure 1 illustrates the XRD patterns of the four prepared (1-x)LiFePO4·xLi3V2(PO4)3 powders A–D. It can be observed the main phase in each sample is LiFePO4 of a well-crystalline olivine-type structure (PDF #40–1499), while some small peaks corresponding to a monoclinic Li3V2(PO4)3 with a P21/n space group (PDF #80–1515) can be detected in sample C and sample D. The content of LVP is too low to be detected in sample B. Furthermore, no other impurity phase can be observed, suggesting that the carbon coating in these samples is either amorphous or too thin to be detected by XRD. The contents of the residual carbon are 1.07 wt% (A), 1.34 wt% (B), 1.63 wt% (C), and 1.63 wt% (D), respectively, according to the results taken from an infrared carbon-sulfur analyzer.

XRD patterns of the four samples (A—a, B—b, C—c, D—d)
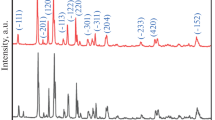
Figure 2 demonstrates the XRD patterns with Rietveld refinement of LFP@C and 0.9LFP·0.1LVP@G, and Table 1 demonstrates the refined unit cell lattice parameters of LFP@C and LFP in 0.9LFP·0.1LVP@G. The reasonably small R factor 9.05 and 10.44% display the convincible refinement results. The lattice parameters in 0.9LFP·0.1LVP decreases slightly in a and b orientations while they increases slightly in c. This result may be attributed to a tiny degree of V doping into LFP, because the ionic radius of V3+ (0.074 nm) is smaller than Fe2+ (0.078 nm). A similar result is reported by Liang et al. in 0.8LFP·0.1LVP [19]. Nevertheless, we notice that the changes in the lattice parameters are very slight so that there is virtually no change in the unit cell volume, i.e., 0.2905(3) vs. 0.2905(2) nm3. It is hard to explain the improvement in rate performance of the 0.9LFP·0.1LVP sample with such a tiny doping effect. As can be seen below, it actually validates another, and more probable, mechanism found in this study, i.e., the difference in the form of carbon coating brought by the LVP modification.

XRD patterns with Rietveld refinement of LFP@C (a) and 0.9LFP·0.1LVP@G (b)
Figure. 3a–d show the SEM images of the four prepared samples. All of them are composed of homogeneous particles with the particle size ranging 150–200 nm. Despite the addition of LVP intermediate powders, there is no obvious increase and aggregation among the particles during the high-temperature heat treatment. Obviously, the presence of carbon can restrain the growth of LFP and LVP particles. Figure. 3e–g show the EDS mapping of sample D (0.9LFP·0.1LVP), which can help to distinguish LFP from LVP particles. To further understand the carbon form and clear morphologies of the primary particles in these samples, the TEM images of baseline LFP (sample A) and 0.9LFP·0.1LVP (sample D) are given in Fig. 4. Figure 4a shows some LFP primary particles and very few scattered free carbon pieces. On the contrary, some transparent graphene-like thin layers are rather visible on the primary particles of 0.9LFP·0.1LVP sample (Fig. 4b). The HRTEM images in Fig. 4b (LFP) and d (0.9LFP·0.1LVP) present that the particles are coated with a uniform amorphous carbon layer, with a thicknesses of about 1.5 and 2 nm, respectively. Under the carbon coatings, both samples show fine lattice fringes with a d-spacing of 0.392 nm, corresponding to the (012) planes of LFP. The observation of the graphene-like thin layers (Fig. 4b) provides a direct evidence that the carbon form changes a lot due to the addition of LVP component. In our previous work on Na3V2(PO4)3, the graphene-decorated Na3V2(PO4)3 material was synthesized through an in situ graphitization process with transition metal oxide VO x as the catalyst. Hence, we believe that the carbon form would change to be more graphene-like to have higher electronic conductivity due to the catalytic function of VO x [27]. During the 750 °C calcination, there must be an in situ generation of graphene on the surfaces of LVP as well as LFP primary particles. Hence, the addition of a small amount content of LVP intermediate powder has a significant impact on the morphology of the particles and particularly the form of their carbon coatings.

SEM images of the four samples (A—a, B—b, C—c, D—d) and EDS mapping of sample D (0.9LFP·0.1LVP) (e–g)

TEM images of LFP@C (sample A) (a) and 0.9LFP·0.1LVP@G (sample D) (b) and HRTEM images of LFP@C (sample A) (c) and 0.9LFP·0.1LVP@G (sample D) (d)
To further identify the form of carbon in the composite, the Raman spectra of the baseline LFP (sample A) and 0.9LFP·0.1LVP (sample D) are presented in Fig. 5. As indicated, two obvious characteristic signals are detected, i.e., the signal at around 1336 cm−1 corresponding to the D band (disorder-induced phonon) and the other signal at around 1597 cm−1 corresponding to the G band (graphite band). Since the relative intensity ratio of D/G cannot give the quantitative result, the D and G bands are sequentially deconvoluted into four peaks (labeled (1)–(4)) by Origin 8.5 software. As shown in Fig. 6, the fitted peaks at around 1350 and 1590 cm−1 correspond to the sp2-type carbon, while the other two fitted peaks at around 1200 and 1500 cm−1 correspond to the sp3-type carbon. The integrated area ratio of sp3 and sp2 (A sp3/A sp2) is related to the content of graphite carbon [28]. For example, a high A sp3/A sp2 ratio indicates that the majority of carbon exists as sp3-type with a low graphitization. Thus, the A sp3/A sp2 ratios of the baseline LFP (Fig. 6a) and 0.9LFP·0.1LVP (Fig. 6b) are calculated to be 0.67 and 0.50, respectively, inferring that a larger amount of carbon exists as sp2-type in 0.9LFP·0.1LVP@G than in LFP@C. The deconvoluted high-resolution XPS C1s spectra for LFP@C and 0.9LFP·0.1LVP@G are shown in Fig. 7, with the lower binding energy corresponding to the sp2-type carbon in the C1s spectrum [29]. The XPS results of the two samples are sequentially deconvoluted into three peaks by using the XPSPEAK 4.1 software, being a mixture of 80% Gaussian and 20% Lorentzian. The deconvoluted peaks locate at about 284, 285, and 286 eV, which correspond to sp2, sp3, and C–O bonds, respectively [30]. The integrated area ratio of sp3 and sp2 peaks are calculated to be 0.63 for LFP and 0.50 for 0.9LFP0.1LVP, which are consistent with above Raman results.

Raman spectra of LFP@C (i.e., x = 0) and 0.9LFP·0.1LVP@G (i.e., x = 0.1)

Deconvoluted Raman spectra of LFP@C (a) and 0.9LFP·0.1LVP@G (b)

Deconvoluted high-resolution XPS C1s spectra for LFP@C (a) and 0.9LFP0.1LVP@G (b)
Figure 8a shows the cyclic voltammograms of the four samples in the second cycle. Obviously, only one couple of redox peaks are observed on the CV curve of LFP (sample A), while four anodic peaks and three cathodic peaks can be clearly observed on the CV curves of samples B–D. The pair peaks of sample A at 3.61/3.26 V are ascribed to the oxidation and reduction of Fe2+/Fe3+ from LFP. Samples B–D all show the redox couple peaks at 3.59/3.30, 3.57/3.30, and 3.56/3.31 V, respectively. The well-defined peaks and smaller values of potential intervals (∆E) between Fe2+/Fe3+ from LFP demonstrates the low resistance in the electrode reactions of these samples. The other three anodic peaks and two cathodic peaks are associated with the V3+/V4+ redox reactions from LVP in samples B to D. Comparing the CV curves of samples B to D with that of sample A, their sharper peaks and smaller ∆E indicate an easier kinetic process for the LVP-modified materials. This can be attributed to the carbon form change from sp3 to sp2 hybridization, being in agreement with the result of TEM and Raman measurement.

The 2nd cycle CV curves of the four samples (1-x)LFP·xLVP (x = 0, 0.02, 0.05, 0.1) at a scanning rate of 0.1 mV s−1 (a) and the initial charge-discharge curves in the voltage range of 2.2–4.3 V for the four samples at 0.1 C (b)
Figure 8b shows the first-cycle charge-discharge profiles for samples A to D. The charge and discharge plateaus are in agreement with the above CV results, and the initial discharge capacity of four sample are 170.6, 170.1, 168.3 and 160.7 mAh g−1 at 0.1 C, respectively. The obvious discharge capacity decrease of sample D is attributed to too much addition of LVP powders (10 wt%). Sample B (2%) and sample C (5%) show slight capacity loss because of less addition of LVP powders.
The cycling behaviors of samples A to D are given in Fig. 9a. The composite electrodes deliver a first-cycle discharge capacity of 161.7, 163.2, 163.6, and 157.8 mAh g−1 at 1 C, respectively. After 100 cycles, their discharge capacity values are 153.6, 154.9, 153.9, and 151.3 mAh g−1, which correspond to a capacity retention of 95, 95, 94.1 and 95.9%, respectively, confirming the good reversibility of the four electrode materials.

a–d The cycling performances of the four samples (1-x)LFP·xLVP (x = 0, 0.02, 0.05, 0.1) at 1 C (a) and their rate performances (b) at room temperature in the voltage ranges of 2.2–4.3 V; rate performances (c), and the discharge curve of the baseline LFP and 0.9LFP·0.1LVP at 0.2 C (d) at − 20 °C in the voltage ranges of 2.2–4.3 V
In the commercially viable systems, the rate performance of electrode materials has significant meaning. Figure 9b indicates the rate capability of samples A to D in the voltage range of 2.2–4.3 V. Obviously, compared with the baseline LFP, the LVP-modified samples (1-x)LFP·xLVP can release much higher discharge capacities especially at high rates (10, 15, and 20 C). The baseline LFP (i.e., sample A) can only deliver 95.5 mAh g−1 at 10 C and almost close to 0 mAh g−1 at 20 C, while other three samples show much better rate performance. Specifically, sample B delivers 137.6 and 103.4 mAh g−1, sample C delivers 140.1 and 112.6 mAh g−1, and sample D delivers 143.6 and 119.2 mAh g−1 at 10 and 20 C, respectively. The most distinguished difference in these samples here is the amount of graphene-like carbon coatings. Compared with other reported LFP samples with different carbon contents prepared by different methods, 0.9LFP·0.1LVP shows an obvious superiority in the rate performance (Table 2). Figure 9c, d also show the comparison of the rate performance at a low temperature of −20 °C for sample A and sample D. Obviously, the low-temperature performance of the LVP-modified sample is also better than that of the baseline LFP.
Figure 10 indicates the EIS spectra in the frequency range of the four samples. It can be observed that they all show a semicircle in the medium-frequency region corresponding to the charge transfer resistance at the interface between the electrodes and the electrolyte. Obviously, the LVP-modified samples, i.e. B–D, give rise to smaller resistance value than the baseline LFP (i.e., sample A). Furthermore, we measured the EIS spectra of LFP and 0.9LFP·0.1LVP before and after 50 cycles as shown in Fig. 11. The sample 0.9LFP·0.1LVP also displays smaller resistance value after 50 cycles. The EIS results demonstrate that the graphene-like carbon coatings indeed result in fast conduction of both electrons and lithium ions in the cells than in the baseline LFP.

Electrochemical impendence spectra of the cells with the four samples (1-x)LFP·xLVP (x = 0, 0.02, 0.05, 0.1) as the cathodes after three cycles of activation at 0.1 C and then controlled to 50% of depth-of-discharge

Electrochemical impendence spectra of the half-cells with LFP and 0.9LFP0.1LVP as the cathodes after three cycles of activation at 0.1 C and then after one and 50 cycles at 1 C. The cell states were all controlled to 50% of depth-of-discharge
Conclusions
We have clarified the primary improvement mechanism of Li3V2(PO4)3-modified LiFePO4 cathode materials. With a small amount of Li3V2(PO4)3 in the synthesis of LiFePO4-based active powders (i.e., LFP·LVP@G), the form of carbon coating on the primary particles changes partially from sp3 to sp2 hybridization in the carbon-carbon covalent bonding. As a cathode material for Li-ion batteries, LFP·LVP@G samples exhibit excellent rate and low-temperature performances. Such a modification synthesis strategy is an effective method that can be extended to other electrode materials with a low electronic conductivity.
References
Padhi AK, Nanjundaswarmy KS, Goodenough JB (1997) Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J Electrochem Soc 144:1188–1194
Okada S, Sawa S, Egashira M, Yamaki JI, Tabuchi M, Kageyama H, Konishi T, Yoshino A (2001) Cathode properties of phospho-olivine LiMPO4 for lithium secondary batteries. J Power Sources 97-98:430–432
Delacourt C, Poizot P, Morcrette M, Tarascon JM, Masquelier C (2004) One-step low-temperature route for the preparation of electrochemically active LiMnPO4 powders. Chem Mater 16:93–99
Yamada A, Hosoya M, Chung SC, Kudo Y, Hinokuma K, Liu KY, Nishi Y (2003) Olivine-type cathodes: achievements and problems. J Power Sources 119-121:232–238
Li G, Azuma H, Tohda M (2002) LiMnPO4 as the cathode for lithium batteries. Electrochem Solid State Lett 5:A135–A137
Yang J, Xu JJ (2006) Synthesis and characterization of carbon-coated lithium transition metal phosphates LiMPO4 (M= Fe, Mn, Co, Ni) prepared via a nonaqueous sol-gel route. J Electrochem Soc 153:A716–A723
Sturaa E, Nicolini C (2006) New nanomaterials for light weight lithium batteries. Anal Chim Acta 568:57–64
Ding B, Xiao PF, Ji G, Ma Y, Lu L, Lee JY (2013) High-performance lithium-ion cathode LiMn0.7Fe0.3PO4/C and the mechanism of performance enhancements through Fe substitution. ACS Appl Mater Interfaces 5:12120–12126
Yuan LX, Wang ZH, Zhang WX, Hu XL, Chen JT, Huang YH, Goodenough JB (2011) Development and challenges of LiFePO4 cathode material for lithium-ion batteries. Energy Environ Sci 4:269–284
Zhao Y, Peng LL, Liu BR, Yu GH (2014) Single-crystalline LiFePO4 nanosheets for high-rate Li-ion batteries. Nano Lett 14:2849–2853
Wang L, He XM, Sun WT, Wang JL, Li YD, Fan SS (2012) Crystal orientation tuning of LiFePO4 nanoplates for high rate lithium battery cathode materials. Nano Lett 12:5632–5636
Liao XZ, Ma ZF, Gong Q, He YS, Pei L, Zeng LJ (2008) Low-temperature performance of LiFePO4/C cathode in a quaternary carbonate-based electrolyte. Electrochem Commun 10:691–694
Wu KP, Hu GR, Du K, Peng ZD, Cao YB (2015) Improved electrochemical properties of LiFePO4/graphene/carbon composite synthesized from FePO4·2H2O/graphene oxide. Ceram Int 41:13867–13871
Zou BK, Wang HY, Qiang ZY, Shao Y, Sun X, Wen ZY (2016) Mixed-carbon-coated LiMn0.4Fe0.6PO4 nanopowders with excellent high rate and low temperature performances for lithium-ion batteries. Electrochim Acta 196:377–385
Naik A, Zhou J, Gao C, Liu GZ, Wang L (2016) Rapid and facile synthesis of Mn doped porous LiFePO4/C from iron carbonyl complex. J Energy Inst 89:21–29
Ban CM, Yin WJ, Tang HW, Wei SH, Yan YF, Dillon AC (2012) A novel codoping approach for enhancing the performance of LiFePO4 cathodes. Adv Energy Mater 2:1028–1032
Ding Y, Jiang Y, Xu F, Yin J, Ren H, Zhuo Q, Long Z, Zhang P (2010) Preparation of nano-structured LiFePO4/graphene composites by co-precipitation method. Electrochem Commun 12:10–13
Li J, Zhang L, Zhang LF, Hao WW, Wang HB, Qu QT (2014) In-situ growth of graphene decorations for high-performance LiFePO4 cathode through solid-state reaction. J Power Sources 249:311–319
Liang SQ, Cao XX, Wang YP, Hu Y, Pan AQ, Cao GZ (2016) Uniform 8LiFePO4·Li3V2(PO4)3/C nanoflakes for high-performance Li-ion batteries. Nano Energy 22:48–58
Cao XX, Pan AQ, Zhang YF, Li JW, Luo ZG, Yang X (2016) Nanorod-Nanoflake Interconnected LiMnPO4·Li3V2(PO4)3/C Composite for High-Rate and Long-Life Lithium-Ion Batteries. ACS Appl Mater Interfaces 8:27632–27641
Guo Y, Huang YD, Jia DZ, Wang XC, Sharma N, Guo ZP (2014) Preparation and electrochemical properties of high-capacity LiFePO4–Li3V2(PO4)3/C composite for lithium-ion batteries. J Power Sources 246:912–917
Zhang JF, Shen C, Zhang B, Zheng JC, Peng CL, Wang XW (2014) Synthesis and performances of 2LiFePO4·Li3V2(PO4)3/C cathode materials via spray drying method with double carbon sources. J Power Sources 267:227–234
Zhong SK, Wu L, Liu JQ (2012) Sol-gel synthesis and electrochemical properties of 9LiFePO4·Li3V2(PO4)3/C composite cathode material for lithium ion batteries. Electrochim Acta 74:8–15
He W, Wei CL, Zhang XD, Wang YY, Liu Q, Shen JX (2016) Li3V2(PO4)3/LiFePO4 composite hollow microspheres for wide voltage lithium ion batteries. Electrochim Acta 219:682–692
Wang L, Li ZC, Xu HJ, Zhang KL (2008) Studies of Li3V2(PO4)3 additives for the LiFePO4-based Li ion batteries. J Phys Chem C 112:308–312
Gao C, Liu H, Liu GB, Zhang J, Wang W (2013) High-rate performance of xLiFePO4·yLi3V2(PO4)3/C composite cathode materials synthesized via polyol process. Mater Sci Eng B 178:272–276
Hu Q, Liao JY, Zou BK, Wang HY, Chen CH (2016) In situ catalytic formation of graphene decoration on Na3V2(PO4)3 particles for ultrafast and long-life sodium storage. J Mater Chem A 4:16801–16804
Zhu ZQ, Cheng FY, Chen J (2013) Investigation of effects of carbon coating on the electrochemical performance of Li4Ti5O12/C nanocomposites. J Mater Chem A 1:9484–9490
Yeganeh M, Coxon PR, Brieva AC (2007) Atomic hydrogen treatment of nanodiamond powder studied with photoemission spectroscopy. Phys Rev B 75:155404
Sotoma S, Akagi K, Hosokawa S (2015) Comprehensive and quantitative analysis for controlling the physical/chemical states and particle properties of nanodiamonds for biological applications. RSC Adv 5:13818–11382
Wang B, Liu TF, Liu AM, Liu GJ, Wang L, Gao TT, Wang DL, Zhao XS (2016) A hierarchical porous C@LiFePO4/Carbon nanotubes microsphere composite for high-rate lithium-ion batteries: combined experimental and theoretical study. Adv Energy Mater:1600426
Hu Y, Gu DW, Jiang HY, Wang L, Sun HS, Wang JP, Shen LJ (2016) Electrochemical performance of LiFePO4/C via coaxial and uniaxial electrospinning method. Adv Chem Eng Sci 6:149–157
Song JJ, Sun B, Liu H, Ma ZP, Chen ZH, Shao GJ, Wang GX (2016) Enhancement of the rate capability of LiFePO4 by a new highly graphitic carbon-coating method. ACS Appl Mater Interfaces 8:15225–15231
Acknowledgements
We would like to show gratitude to the National Science Foundation of China (grant no. 51577175) and NSAF (grant no. U1630106) for its financial support. We also thank Elementec Ltd. in Suzhou for its technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hu, Q., Liao, JY., Zou, BK. et al. Improving the rate and low-temperature performance of LiFePO4 by tailoring the form of carbon coating from amorphous to graphene-like. J Solid State Electrochem 22, 797–805 (2018). https://doi.org/10.1007/s10008-017-3777-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-017-3777-y