
Abstract
The nano-particles, Li3V2(PO4)3 embedded in carbon matrix, are synthesized by a novel two-step procedure based on a sol–gel method. In the two-step process, the carbon-encapsulated trivalent vanadium phosphorus precursors are obtained without adding lithium compound in the first step. Because of carbon matrix, small particle size of Li3V2(PO4)3/C composite is obtained in the second step, which involves lithiation and heating to 800 °C in argon. The electronic conductivity of Li3V2(PO4)3/C composite is 2.88 × 10−3 S cm−1, increased by five orders of magnitude compared with the pristine Li3V2(PO4)3 (2.3 × 10−8 S cm−1). The prepared Li3V2(PO4)3/C shows high discharge capacity 132 mAh g−1 (3.0–4.3 V), which is up to 99.2 % of the theoretical value, and good high-rate specific discharge capacity, which is greater than 100 mAh g−1 at a rate of 10 C. Even after cycling at 15 C, the charge–discharge performance of the prepared Li3V2(PO4)3/C cathode hardly deteriorates at 1 C indicating that the structure of Li3V2(PO4)3 is not destroyed irreversibly at high rate. The improved electrochemical properties of the Li3V2(PO4)3/C cathode electrodes are also verified by electrochemical impedance spectroscopy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Enhanced energy density, power density, cyclability, and safety of lithium-ion batteries have been the research objects since it became commercialized in 1990. In short, it is to get better performance. These new needs have boosted research and optimization of new materials for lithium-ion batteries. NASICON monoclinic Li3V2(PO4)3 phase, a cathode material that, besides having a high theoretical specific capacity (197 mAh g−1, 3.0–4.8 V; 133 mAh g−1, 3.0–4.3 V) and stable crystalline structure displaying remarkable rate performance, will apply to vehicle batteries especially [1–3]. The host lattice is composed of interconnected corner-shared P-O tetrahedron and V-O octahedron, which forms a 3D framework. The three Li sites account for a halfway situation vis-a-vis the two adjacent members [4–6]. Because V-O octahedron is separated by P-O tetrahedron, Li3V2(PO4)3 presents poor solid state diffusion and electrical conductivity, discharge specific capacity is low at high rate charge [7, 8]. Li3V2(PO4)3 nano-particles coated with carbon layers are obtained in our work resulting in improved ion and electronic conductivity.
The small size and large surface area of nano-materials provide such greater contact area between the electrode material and the electrolyte. The distance of lithium ions diffusing across the electrode is shortened. Therefore, improved charge/discharge ability, that is, a higher rate capability, can be expected for nano-structured electrodes. The fabrication of nano-structured electrodes has become one of the main goals in battery materials [9]. Apart from the sol–gel preparative methods [10–13], a wide variety of synthetic approaches have been developed to obtain nano-particles and to improve the rate capabilities of the Li3V2(PO4)3 materials, such as solid state method [14–16], microwave method [17], hydrothermal method [18, 19], and electrostatic spray deposition [20]. However, it is known from ceramic processing [21] that light elements, such as lithium, can increase the particle growth rate at high temperatures, which is due to the increased diffusion rate of the light elements. To address these defects, Pivko etc. synthesized LiMnPO4 with small particle size (15–20 nm) via a two-step technique [22]. Barker etc. synthesized LiVPO4F by a novel two-step reaction method based on a carbon thermal reduction (CTR) process [23].
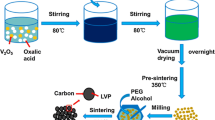
In order to obtain the nano particle size of LVP/C composite, we herein propose a two-step process based on the sol–gel method to prepare Li3V2(PO4)3. In this article, we present a novel approach toward a two-step synthesis of small particles, Li3V2(PO4)3, embedded in a carbon matrix via an oxalic acid-based sol–gel process assisted by glucose, in which oxalic acid and glucose serve as double carbon sources. To achieve this goal, we modified the first step (sol–gel) without adding lithium compounds into solution. Then, the prepared trivalent vanadium phosphorus composites (VPO) were mixed lithium salts by using a planetary ball milling. Homogeneous Li3V2(PO4)3 particles were distributed in a conductive carbon matrix after the second heat treatment. The electrochemical performances of Li3V2(PO4)3/C particles were investigated.
Experimental
Two-step synthesis
In the first step, oxalic acid (4.40 g) and V2O5 (1.82 g) in a stoichiometric molar ratio 3:1 were dissolved in 30-ml deionized water with magnetic stirring at 80 °C. After a clear blue solution formed, a mixture of stoichiometric NH4H2PO4 (3.45 g) and glucose (1.60 g) was added to the solution while stirring for 30 min (the molar ratio of V:P was 2:3), and dried sol formed in an air oven at 110 °C. The dried sol was pre-decomposed at 350 °C for 5 h, then thermally treated at 800 °C for 2 h in an argon atmosphere.
In the second step, the composite from the first step was mixed with adequate quantity of LiOH (1.40 g) using planetary ball milling for 30 min. The final Li3V2(PO4)3/C composite material was obtained with additional thermal treatment at 800 °C in argon for 12 h (LVP2 for short).
Sol–gel method
A homogeneous mixture of reactants was prepared by stirring stoichiometric quantities of oxalic acid (4.40 g), V2O5 (1.82 g), NH4H2PO4 (3.45 g), LiOH (1.40 g), and glucose (1.60 g) in 30-ml deionized water (the molar ratio of Li:V:P was 3:2:3). The dried sol was pre-decomposed at 350 °C for 5 h, then thermally treated at 800 °C for 12 h in an argon atmosphere. The obtained final Li3V2(PO4)3/C composites was called LVP1 for short. In the sol–gel process, oxalic acid is often used as chelating ligand, which can also act as carbon source and reducing agent. In this work, LVP/C composites were synthesized via oxalic acid-based sol–gel route assisted by glucose. Here, glucose served as the additional carbon source to modify the carbon-coated layer achieved by oxalic acid in LVP/C composites.
The structural properties of the prepared material and the intermediate precursor obtained from the both methods were determined by X-ray diffraction (XRD), using a SIMENS D5000 diffractometer (λ = 1.5406 Å). The surface morphology of the prepared composites was characterized by a field-emission scanning electron microscopy (FE-SEM) (Model JSM-6700F, 5 kV). The morphology of individual nano-particles was examined in a Model JEM-3010 F, 200 kV, transmission electron microscopy (TEM). The TGA−DTA measurements were performed under 20 ml min−1 argon flow at a heating rate of 10 °C min−1 with a simultaneous thermal analysis system (Setsys Evo24, SETARAM, France). The electronic conductivity of powder was determined by two electrode method on a GM-II resistivity tester connected to a 34401A type Digit multimeter.
The positive electrodes comprised weight of 80 % active materials, 10 % carbon black, and 10 % PVDF. These components were mixed in N-methyl-2-pyrrolidone (NMP). The obtained slurry was cast onto a circular aluminum foil with a diameter of 16 mm. Before being transferred to a glovebox (water content <1 ppm), the electrodes were dried in vacuum oven at 120 °C for 12 h. Galvanostatic charge/discharge tests were performed in a coin battery between 3.0–4.3 V (the electrolyte may deteriorate partly when charged to 4.8 V), where the working electrode and counter electrode (lithium foil) were separated by a separator soaked in the electrolyte. The electrolyte consisted of 1 M solution of LiPF6 in a mixture of 1:1 by weight of ethylene carbonate (EC) and dimethyl carbonate (DMC). The galvanostatic charge/discharge tests were performed with an Arbin BT2000 battery tester at different current densities at 25 °C. Rate performance was obtained by applying varying current densities during charge/discharge without additional constant voltage charge (1 C = 133 mA g−1, 3.0–4.3 V). Electrochemical impedance spectroscopy (EIS) was recorded on an electrochemical workstation with the frequency ranging from 10 kHz to 10 mHz and an AC signal of 5 mV in amplitude as the perturbation at 25 °C (CHI660C, Shanghai).
Results and discussion
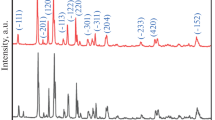
The XRD patterns of Li3V2(PO4)3/C obtained from one-step sol–gel (short for LVP1) and Li3V2(PO4)3/C obtained from two-step approach (short for LVP2) are shown in Fig. 1. All diffraction peaks can be assigned to the single-phase of Li3V2(PO4)3 (JCPDS:97-016-1335) [1, 24]. There is no significant difference between the XRD patterns of LVP1 and those of LVP2. Additionally, no peaks of impurities were detected from the XRD patterns. Pyrolytic carbon obtained from glucose under thermal condition is amorphous and cannot be detected through XRD. The mean crystallite size of LVP1 and LVP2 particles are calculated from broadening of XRD peaks by Scherrer’s equation: D = 0.89λ / (β cosθ), where λ is the X-ray wavelength and β is the full width at half maximum of the diffraction peak on a 2θ scale. Based on diffraction planes of (121), (020) and (220), the mean value of grain size of LVP1 and LVP2 are 31 and 38 nm, respectively.

XRD patterns of LVP1 (a) and LVP2 (b)
In the second step of the synthesis, lithium salts must be distributed very homogeneously across the precursors to achieve a complete transformation of VPO into Li3V2(PO4)3. The method of ball milling in this work is adopted to achieve the goal of sufficient mixing. As described by Pivko etc., the lithium diffusion through the mixture facilitates the transformation of VPO into Li3V2(PO4)3 under high heating temperature [22]. After heating mixtures, a sample is obtained without detectable impurities (Fig. 1b).
To better understand the formation mechanism, structure, and particle morphology of the intermediate phase VPO, we performed TGA-DTA analyzation of precursors, XRD and SEM characterization of VPO samples sintered at 800 °C. The TGA-DTA analytical curves of mixed precursors are shown in Fig. 2a. The inset shows a DTA curve of glucose. Corresponding to the peaks of glucose, the first main endothermic peak occurs at around 180 °C in the DTA curve of precursors. The first part of its weight loss attributes to the early decomposition and dehydration of glucose. The large endothermic peak at 280 °C and the major weight loss can be attributed to the formation of VPO compounds with the consequent formation of NH3, H2O, and CO2 gas caused by decomposition [25]. XRD pattern of VPO is shown in Fig. 2b, where there are no obvious peaks and no indication of the presence of NH4H2PO4 or VPO4 samples. The VPO samples formed in the first step of synthesis are amorphous phase, and there are no VPO4 crystals generated from the reaction. Figure 2c shows the SEM micrograph of the intermediate phase VPO. As can be seen, flake structure appears in the morphology of the VPO. As shown in XRD (Fig. 1), the VPO completely transformed into Li3V2(PO4)3 crystal (LVP2) ultimately after the addition of lithium salts and high temperature heating.

a TGA-DTA results of the precursor mixture. The inset shows a DTA curve of glucose. b XRD pattern. c SEM micrograph of the VPO
Figure 3a, b shows the SEM images of the LVP1 and LVP2 composite. It can be observed that the morphology of both LVP1 and LVP2 particles is irregular. The morphology of both LVP1 and LVP2 particles are different from that of the VPO, indicating Li atoms played an important role in the formation of LVP/C composites.

SEM micrographs of LVP1 (a) and LVP2 (b)
Figure 4a, b shows the low magnification TEM images of LVP1 and LVP2, respectively. As can be seen, there is more carbon matrix existing in LVP2 particles than in LVP1 particles. Meanwhile, the particle size of LVP2 (<300 nm) is much smaller than of LVP1 (ca. 500 nm) suggesting that Li atoms facilitated formation of large particle size under high temperature, in accordance with those reports described in the “Introduction” part. The matrix formation of carbon appears in the interstitial grain-boundary region, which can limit the growth of particles and provide good contact between the particles. The crystal nature of Li3V2(PO4)3 is evidenced by the SAED patterns (inset of Fig. 4c, d). As can be seen, LVP1 particles are composed of small polycrystalline grain, whereas LVP2 particles are more like monocrystals with large size. These results are in good agreement with the XRD investigations. The higher degree of crystallinity of LVP2 than that of LVP1 may contribute to the faster lithium ion transport, which is the neck of a bottle, when charging at large current density. By observing the edge of one particle, we could also find the presence of carbon layer, as seen in Fig. 4c, d. The distribution of amorphous carbon around particles is continuous, but the thickness of carbon coating is not completely homogeneous. The thickness of amorphous carbon coating can be controlled by changing the weight of pyrolytic carbon source [26, 27]. The residual carbon content is approximately 4.75 % wt. in LVP1 and the same as in LVP2. The residual carbon content of glucose is determined by pyrolysis in Ar atmosphere from room temperature to 800 °C in tubular furnace.

TEM micrographs of LVP1 (a, c) and LVP2 (b, d) at different magnification. Insets in (c) and (d) are the corresponding SAED patterns of LVP1 and LVP2 in red square areas, respectively
The electronic conductivity of the as-prepared LVP1 and LVP2 composite is 1.38 × 10−3 S/cm and 2.88 × 10−3 S/cm, respectively, which are much higher than the pristine Li3V2(PO4)3 (2.3 × 10−8 S/cm) at 300 K [5]. According to the TEM results, higher conductivity of LVP2 than that of LVP1 is attributed to the small particle size and carbon matrix in the composite. In general, one could come to a conclusion: the particle size of active particles can be suppressed by simultaneously introducing carbon through carbon precursors and avoiding lithium during the first firing step. The Li3V2(PO4)3 particles imbedded in the carbon matrix can be obtained through a two-step routine, which results in improved electronic conductivity.
The discharge curves for LVP1 cells and LVP2 cells at different current densities are shown in Fig. 5, which are measured in the potential range of 3.0–4.3 V at 25 °C. All cells exhibited three discharge plateaus around 4.0, 3.7, and 3.6 V at rate of 0.2 C, which are identified as the two-phase transition processes during the electrochemical reactions, LiV2(PO4)3 → Li2V2(PO4)3 → Li2.5 V2(PO4)3 → Li3V2(PO4)3 [5, 7]. The discharge capacities for LVP1 and LVP2 are 131 mAh g−1 and 132 mAh g−1, accounting for 98.5 % and 99.2 % of theoretical specific capacity (133 mAh g−1) respectively. When the rates increased, namely the charge–discharge current density increased, the discharge plateaus evolved into a slope gradually, which can be attributed to the electrode polarization [28, 29]. The discharge specific capacities of LVP1 cells are much smaller than those of LVP2 cells at increasing rates, due to the deteriorating polarization degree of LVP1 cells. The superior charge–discharge performance of LVP2 is attributed to the decreased particle size and improved electronic conductivity, in good agreement with TEM investigations above.

Discharge curves of LVP1 cells (a) and LVP2 cells (b) at various rates
The rate capacities of LVP1 and LVP2 samples are shown in Fig. 6a. The electrodes were cycled between 3.0 and 4.3 V versus metallic lithium without additional constant voltage treatment. It is obvious that the high-rate performance of LVP2 cells is better than that of LVP1. The discharge capacities of LVP1 and LVP2 samples are almost the same at 0.2, 1, and 5 C (132, 125, and 115 mAh g−1). The conductivity capability and particle size have little impact on the discharge capacities of Li3V2(PO4)3 at low charge–discharge rate. However, when the charge–discharge rates rise to 10 and 15 C, the discharge capacities of LVP1 are about 80 and 60 mAh g−1, which is much smaller than those of LVP2, 100 and 88 mAh g−1, respectively. The inset of Fig. 6a shows the charge–discharge curve of LVP2 electrodes at 1 C after cycling 5 times at 15 C. The apparent plateaus are observed and discharge capacities hardly deteriorate yet. The discharge capabilities do not decrease even if LVP2 cycles at high current density charge–discharge rate. This may be ascribed to the decreasing particle size and improved conductivity of LVP2. Because the smaller particle size leads to shorter Li ions transport distance, it facilitates the fast ion transport rate. Also, the benefit of Li3V2(PO4)3 particles imbedded in the carbon matrix in the electrode leads to enhanced rate performance of samples. Cycle performance of LVP1 and LVP2 at a rate of 10 C is shown in Fig. 6b. Both LVP1 and LVP2 show good cycling stability after 50 cycle numbers. As shown in the previous results, LVP2 exhibits larger capacity than LVP1 because of superior structure. We can conclude that Li3V2(PO4)3 samples prepared by two-step process are in favor of superior rate and cycling performance, due to decreased particle size and enhanced electronic conductivity of the LVP2 composite.

a Discharge capacities of LVP1 cells and LVP2 cells at various rates. Inset shows the charge–discharge curves of LVP2 cells at a rate of 1 C after cycling at 15 C. b Cycle performance of LVP1 and LVP2 at rate of 10 C
The electrochemical impedance spectroscopy (EIS) measurements were performed for LVP1 and LVP2 electrodes at the potential of 3.0 V after the first cycle at a 0.2 C rate and the Nyquist plots are shown in Fig. 7. Both electrodes exhibited a depressed semicircle in the high-frequency region and an inclined line in the low-frequency region. The high-frequency semicircle is assigned to the charge-transfer impedance in the electrode/electrolyte interface, and the inclined line corresponds to the lithium-ion diffusion process [30]. It is known that the electrolyte resistance through the separator and contact resistance contribute to a large fraction of the Ohmic potential drop while the charge transfer resistance may control the rate of lithium insertion. The charge-transfer impedances for LVP1 and LVP2 electrodes are 115 and 60 Ω cm−2, respectively. The charge transfer impedances decreased greatly with the decreased particle size and enhanced electronic conductivity because of decreased Li-ion transport distance and fast ion transport rate. Moreover, the lower contact resistance of LVP2 than that of LVP1 electrodes led to enhanced rate performance, in accordance with the above results (Figs. 5 and 6).

Nyquist plots of LVP1 and LVP2 electrodes at potential of 3.0 V after initial charge–discharge cycle
Conclusion
The Li3V2(PO4)3 particles embedded in a carbon matrix were obtained via a two-step approach. The particle size of active particles can be suppressed by simultaneously introducing carbon coating through carbon precursors and avoiding lithium during the first firing step. The LVP2 electrodes exhibited enhanced discharge capacities, 25 % higher than those of LVP1 electrodes at a rate of 10 C or higher rate. The LVP2 electrodes exhibited decreased charge transfer impedances and enhanced rate performance, which are attributed to the decreased particle size and enhanced electronic conductivity of the material. The primary results illustrate that the as-prepared Li3V2(PO4)3/C can be a promising cathode material for higher energy density and power demanding lithium-ion batteries.
References
Gaubicher J, Wurm C, Goward G, Masquelier C, Nazar L (2000) Rhombohedral form of Li3V2(PO4)(3) as a cathode in Li-ion batteries. Chem Mater 12:3240, − +
Cahill LS, Chapman RP, Britten JF, Goward GR (2006) (7)Li NMR and two-dimensional exchange study of lithium dynamics in monoclinic Li(3)V(2)(PO(4))(3. J Phys Chem B 110:7171–7177
Cheng B, Zhang X-D, Ma X-H, Wen J-W, Yu Y, Chen C-H (2014) Nano-Li3V2(PO4)3 enwrapped into reduced graphene oxide sheets for lithium-ion batteries. J Power Sources 265:104–109
Saı̈di MY, Barker J, Huang H, Swoyer JL, Adamson G (2003) Performance characteristics of lithium vanadium phosphate as a cathode material for lithium-ion batteries. J Power Sources 119–121:266–272
Yin SC, Strobel PS, Grondey H, Nazar LF (2004) Li2.5V2(PO4)3: a room-temperature analogue to the fast-ion conducting high-temperature γ-phase of Li3V2(PO4)3. Chem Mater 16:1456–1465
Burba CM, Frech R (2007) Vibrational spectroscopic studies of monoclinic and rhombohedral Li3V2(PO4)(3). Solid State Ionics 177:3445–3454
Yin SC, Grondey H, Strobel P, Anne M, Nazar LF (2003) Electrochemical property: Structure relationships in monoclinic Li(3-y)V2(PO4)3. J Am Chem Soc 125:10402–10411
Liu HD, Yang G, Zhang XF, Gao P, Wang L, Fang JH et al (2012) Kinetics of conventional carbon coated-Li3V2(PO4)(3) and nanocomposite Li3V2(PO4)(3)/graphene as cathode materials for lithium ion batteries. J Mater Chem 22:11039–11047
Zhang WK, Zhao B, Xia Y, Zhou XZ, Huang H, Zeng HJ et al (2012) One-step synthesis of asphalt based Li3V2(PO4)(3)/C nanocomposites as cathode materials for lithium-ion batteries. Advances in Chemical Engineering, Pts 1–3 396–398:1748–1754
Zhu XJ, Liu YX, Geng LM, Chen LB (2008) Synthesis and performance of lithium vanadium phosphate as cathode materials for lithium ion batteries by a sol–gel method. J Power Sources 184:578–582
Liu H, Gao P, Fang J, Yang G (2011) Li3V2(PO4)3/graphene nanocomposites as cathode material for lithium ion batteries. Chem Commun 47:9110–9112
Zhang L, Xiang H, Li Z, Wang H (2012) Porous Li3V2(PO4)3/C cathode with extremely high-rate capacity prepared by a sol–gel-combustion method for fast charging and discharging. J Power Sources 203:121–125
Yang Y, Xu W, Guo R, Liu L, Wang S, Xie D et al (2014) Synthesis and electrochemical properties of Zn-doped, carbon coated lithium vanadium phosphate cathode materials for lithium-ion batteries. J Power Sources 269:15–23
Wang LJ, Zhou XC, Guo YL (2010) Synthesis and performance of carbon-coated Li3V2(PO4)(3) cathode materials by a low temperature solid-state reaction. J Power Sources 195:2844–2850
Wang JW, Liu J, Yang GL, Zhang XF, Yan XD, Pan XM et al (2009) Electrochemical performance of Li3V2(PO4)(3)/C cathode material using a novel carbon source. Electrochim Acta 54:6451–6454
Rui XH, Li C, Chen CH (2009) Synthesis and characterization of carbon-coated Li3V2(PO4)(3) cathode materials with different carbon sources. Electrochim Acta 54:3374–3380
Yang G, Liu HD, Ji HM, Chen ZZ, Jiang XF (2010) Temperature-controlled microwave solid-state synthesis of Li3V2(PO4)(3) as cathode materials for lithium batteries. J Power Sources 195:5374–5378
Sun C, Rajasekhara S, Dong Y, Goodenough JB (2011) Hydrothermal synthesis and electrochemical properties of Li3V2(PO4)3/C-based composites for lithium-ion batteries. ACS Appl Mater Interfaces 3:3772–3776
Chang CX, Xiang JF, Shi XX, Han XY, Yuan LJ, Sun JT (2008) Hydrothermal synthesis of carbon-coated lithium vanadium phosphate. Electrochim Acta 54:623–627
Wang L, Zhang LC, Lieberwirth I, Xu HW, Chen CH (2010) A Li3V2(PO4)(3)/C thin film with high rate capability as a cathode material for lithium-ion batteries. Electrochem Commun 12:52–55
Nicholas JD, De Jonghe LC (2007) Prediction and evaluation of sintering aids for cerium gadolinium oxide. Solid State Ionics 178:1187–1194
Pivko M, Bele M, Tchernychova E, Logar NZ, Dominko R, Gaberscek M (2012) Synthesis of nanometric LiMnPO4via a two-step technique. Chem Mater 24:1041–1047
Barker J, Saidi MY, Swoyer JL (2003) Electrochemical insertion properties of the novel lithium vanadium fluorophosphate, LiVPO4 F. J Electrochem Soc 150:A1394–A1398
Rui XH, Yesibolati N, Chen CH (2011) Li3V2(PO4)(3)/C composite as an intercalation-type anode material for lithium-ion batteries. J Power Sources 196:2279–2282
Allen CJ, Jia QY, Chinnasamy CN, Mukerjee S, Abraham KM (2011) Synthesis, structure and electrochemistry of lithium vanadium phosphate cathode materials. J Electrochem Soc 158:A1250–A1259
Zhang L-L, Li Y, Peng G, Wang Z-H, Ma J, Zhang W-X et al (2012) High-performance Li3V2(PO4)3/C cathode materials prepared via a sol–gel route with double carbon sources. J Alloys Compd 513:414–419
Qiao YQ, Wang XL, Xiang JY, Zhang D, Liu WL, Tu JP (2011) Electrochemical performance of Li3V2(PO4)(3)/C cathode materials using stearic acid as a carbon source. Electrochim Acta 56:2269–2275
Wang ZX, Liu LJ, Chen LQ, Huang XJ (2002) Structural and electrochemical characterizations of surface-modified LiCoO2 cathode materials for Li-ion batteries. Solid State Ionics 148:335–342
Choi D, Wang D, Bae IT, Xiao J, Nie Z, Wang W et al (2010) LiMnPO4 nanoplate grown via solid-state reaction in molten hydrocarbon for Li-ion battery cathode. Nano Lett 10:2799–2805
Thorat IV, Joshi T, Zaghib K, Harb JN, Wheeler DR (2011) Understanding rate-limiting mechanisms in LiFePO4 cathodes for Li-Ion batteries. J Electrochem Soc 158:A1185–A1193
Acknowledgments
This work was supported by the National Science Foundation of China (Grant No. 51172068 and Grant No. 51372079) and the Industry Support Program of Hunan Province (No. 2014GK3102).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, X., Han, S. & Fan, C. Synthesis of Li3V2(PO4)3/C with enhanced rate performance by a two-step technique. Ionics 21, 1509–1515 (2015). https://doi.org/10.1007/s11581-014-1335-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-014-1335-5