
Abstract
Carbon-supported Ni@Ag core-shell nanoparticles were synthesized and used as the anode electrocatalyst for direct borohydride-hydrogen peroxide fuel cell (DBHFC). The morphology, structure, and composition of the as-prepared electrocatalysts are characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD), and energy dispersive spectroscopy (EDS). Electrochemical characterizations are performed by cyclic voltammetry (CV), chronoamperometry (CA), linear scan voltammetry with rotating disk electrode (LSV RDE), and fuel cell test. The catalytic behaviors and main kinetic parameters (e.g., Tafel slope, number of electrons exchanged, exchange current density, and apparent activation energy) toward BH ‐4 oxidation on Ag/C and Ni@Ag/C electrocatalysts are determined. Results show that the as-prepared nanoparticles have a core-shell structure with the average size approximately 13 nm. The kinetics of NaBH4 oxidation is faster for Ni@Ag/C than that for Ag/C. Among the as-prepared catalysts, the highest transition electron value and the lowest apparent activation energy are obtained on Ni1@Ag1/C; the values are 4.8 and 20.23 kJ mol−1, respectively. The DBHFC using Ni1@Ag1/C as anode electrocatalyst and Pt mesh (1 cm2) as cathode electrode obtains the maximum anodic power density as high as 8.54 mW cm−2 at a discharge current density of 8.42 mA cm−2 at 25 °C.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Because of the depletion of fossil fuel resources and ever-increasing environmental pollution, the use of clean energy has become an irreversible trend. Compared with other power sources, fuel cells have attracted considerable attention due to their ability to provide an efficient and harmless environment way of energy generation. Direct liquid fuel cells, fed with borohydride, methanol [1], or ethylene glycol [2], have been studied as a substitute to hydrogen for low-temperature fuel cells. Although both direct borohydride fuel cell (DBFC) and direct methanol fuel cell (DMFC) can solve the problems of hydrogen storage, DBFC supersedes DMFC in terms of capacity value, electrochemical activity, theoretical open circuit voltage, and power performance at ambient temperature [3]. In addition, DBFC uses alkaline solution as the fuel which has relatively low corrosion activity, and therefore, it is possible to apply readily available and low-cost non-precious metal anode catalysts. Besides oxygen, hydrogen peroxide can also be used as the oxidant in DBFC. The reactions are as follows:
Obviously, direct borohydride-hydrogen peroxide fuel cell (DBHFC), which uses borohydride in alkaline electrolyte as fuel and hydrogen peroxide in acid as oxidant, can yield a theoretical cell voltage as high as 3.01 V. However, the borohydride oxidation reaction (BOR) pathway occurs depending on the anode catalyst material, solution composition, temperature, and so on [4–7]. Usually, a quasi-spontaneous heterogeneous hydrolysis occurs in competition with the eight-electron borohydride oxidation reaction. This hydrolysis apparently proceeds as follows [8]:
In the past few years, various metals have been studied as the anode electrocatalysts for DBFC, such as Pt [9–11], Pd [12, 13], Ni [14, 15], Cu [16], Co [17, 18] known as “catalytic electrodes,” and Au [4, 5, 19, 20], Ag [4, 10, 11, 21], known as “non-catalytic electrodes.” Although Ag exhibits excellent activity toward the oxidation of borohydride and is presented as a non-catalytic material with respect to BH ‐4 hydrolysis, the slow electrode kinetics and low power densities indicate that Ag cannot be used alone [11, 22]. Thus, it is essential to design new and effective Ag-based composite catalysts with superior performance.
The combination of noble metals with other non-noble metals has been developed to make a bimetal nanocomposite, which gives rise to a significant increase in a catalytic performance compared with single-component materials. Among the non-precious metals, nickel has received a lot of attention as catalyst for electrooxidation of BH ‐4 . High performance has been achieved by DBFC using Ni-based composite catalysts [23–27]. Wang et al. [26] reported that the higher electrocatalytic activity toward BOR of the PtNi(7:3)/C could be attributed to the alloy effect and the Pt electronic structure change due to the addition of Ni. Šljukić et al. [27] compared the BOR catalytic activities of Pt/C and Pt0.75M0.25/C (M=Ni or Co), and the electrochemical studies revealed beneficial effect of the Ni metal presence on Pt alloy performances for BH ‐4 oxidation, as higher current densities, higher number of electrons exchanged, and lower activation energies were obtained at bimetallic Pt alloy electrocatalysts compared to pure Pt. Therefore, kinds of effective composition of Ag and Ni are suggested. Feng RX [28] prepared AgNi alloy by mechanical ball milling method, and the results showed that the AgNi-catalyzed borohydride fuel cells exhibited a higher discharge voltage and capacity compared with Ag and the borohydride oxidation on AgNi surface could transfer 7.4 electrons.
Especially in recent years, nano-sized core-shell bimetallic particles have received much attention due to their superior optical, electronic, catalytic, and magnetic properties compared to their monometallic counterparts [29]. These interesting physicochemical properties result from the combination of two kinds of metals and their fine structures, evolving new surface characteristics [30]. Many kinds of core-shell nanoparticles were prepared and used as electrocatalysts for fuel cells [31–34]. In particular, we prepared Ni@Au/C and Cu@Ag/C core-shell nanopaticles which were used as anode electrocatalysts in DBHFC, and the results indicated that the Ni@Au/C and Cu@Ag/C catalysts exhibit higher catalytic activity for the direct electrooxidation of borohydride compared with Au/C or Ag/C [35, 36]. Besides, Baletto et al. [37] found that, in molecular dynamics simulations of the growth of bimetallic AgNi clusters, AgNi clusters could prefer to form a core-shell structure because this compound shows a strong tendency to phase separation in the bulk with Ag segregating at the surface. Thus, evaluation of the catalytic activities for Ni@Ag/C core-shell catalysts toward BH ‐4 oxidation is valuable.
In this paper, carbon-supported Ni@Ag nanoparticles are prepared by the impregnation reduction method in aqueous solution and used as the anode electrocatalyst for DBHFC. The electrochemical properties were investigated by cyclic voltammetry (CV), chronoamperometry (CA), fuel cell test, liner scan voltammetry (LSV) with different electrode rotation speed, temperature, and NaBH4 concentrations to analyze the catalytic activity and kinetics of core-shell nanoparticles toward BH ‐4 oxidation.
Experimental
Ag/C nanopaticles were prepared as follows: 0.0786 g AgNO3 (Chemical Reagent, Tianjin) and 5 mg PVP (Wind boat, Tianjin) dissolved in 10 ml doubly distilled water, and then, this solution was ultrasonically dispersed for 30 min. While dispersing, 10-ml reducing agent containing 1 M NaOH (Sigma-Aldrich, 99 %)+ 2 M NaBH4 (Sigma-Aldrich, 99 %) was added to the solution dropwise under nitrogen atmosphere. After 30 min, 0.2 g XC-72R (Cabot Corp., 250 m2 g−1) was added into the solution and before dispersing for another 30 min. Then, the resultant nanoparticles were separated by centrifugation and washed twice with water and ethanol. These procedures were performed at 25 °C. Finally, the powder was dried at 80 °C for 12 h in a vacuum and then cooled naturally to 25 °C. As a result, the Ag/C catalyst was obtained. Ni@Ag/C nanopaticles were prepared as follows (molar ratio of Ni to Ag is 1:1): 0.0713 g NiCl2·6H2O (Sigma-Aldrich, 99 %), and 5 mg PVP was dissolved in 10 ml doubly distilled water; then, this solution was ultrasonically dispersed for 30 min. While dispersing, 10-ml reducing agent containing 1 M NaOH + 2 M NaBH4 was added to the solution dropwise under nitrogen atmosphere. After 30 min, the Ni ions were reduced completely, and then, the solution composed of 0.051 g AgNO3 and 10 ml doubly distilled water was added dropwise under nitrogen atmosphere. After reacting completely, 0.2 g XC-72R was added into the above solution and dispersed for 30 min. Then, the resultant nanoparticles were separated by centrifugation and washed twice with water and ethanol. The procedures above were performed at 25 °C. Finally, the powder was dried at 80 °C for 12 h in a vacuum and then cooled naturally to 25 °C. As a result, the Ni@Ag/C catalyst was obtained and marked as Ni1@Ag1/C. According to the preceding method, Ni@Ag/C catalysts of different molar ratios of Ni to Ag were prepared as the above method and marked as Ni1@Ag1.5/C, Ni1.5@Ag1/C, and Ni2@Ag1/C. The loading amount of metals in all catalysts is 20 wt.% of total catalyst weight.
Glassy carbon is the basic material of working electrode. Before using, it was wiped by Al2O3 suspension then ultrasonic washed three times in absolute alcohol and distilled water, respectively, to remove the impurities. A total of 10 mg Ag/C or Ni@Ag/C powers were ultrasonically dispersed for 1 h in 1-ml blend solution containing 0.95 ml of ethanol and 0.05 ml of Nafion (5 wt.%). Then, 5 μl of suspension was carefully applied on glassy carbon electrode surface. The dispersed Ag/C or Ni@Ag/C electrocatalyst on the glassy carbon surface was dried at 70 °C. The loading mass of Ag/C or Ni@Ag/C electrocatalyst was 0.25 mg cm−2.
The structure and morphology of the prepared electrocatalysts were examined by transmission electron microscopy (TEM), using JEM-2010 microscope at 200 kV. The Ni-Ag atomic ratio of Ni@Ag/C was determined by energy dispersive spectra (EDS) analysis. X-ray diffractometer (D/max 2500) was employed with Cu Kα radiation (λ = 0.154056 nm) and a graphite monochromator at 40 kV, 100 mA to obtain X-ray diffraction (XRD) patterns of the samples. The 2θ angular regions between 10° and 80° were explored at a scan rate of 8° min−1.
Electrochemical characterization was performed on a standard three-electrode cell using an AFMSRXIE-536 rotating disk electrode (PAR Company, USA) connected to a VMPIII multichannel potentiostat. Pt net (50 meshes, 1 cm2) was used as counter electrode, and Hg/HgO (1 M NaOH) was used as the reference electrode. The cyclic voltammograms were taken at a scan rate of 50 mV s−1, and the potential range was varied between −0.9 and 0.7 V (vs MOE) Ag/C. The CA was taken at 0.2 V and lasted for 60 s. The linear scan voltammograms (LSVs) were taken using a rotating disk electrode at a scan rate of 3 mV s−1. CV, CA, and linear sweep voltammetry were tested in solution composed of 2 M NaOH + 0.1 M NaBH4.
The performance of the direct NaBH4/H2O2 fuel cell was evaluated by a homemade electrochemical testing system at 25 °C. A schematic diagram of the experimental setup was shown in Fig. 1. A compartment cell was used in which the activated NRE-212 cation membrane compressed between the anode and cathode compartment (3 cm × 4 cm × 5 cm). The cell composed of the Ni@Ag/C working electrode, a Pt net (1 cm × 1 cm) as counter electrode and Hg/HgO (1 M NaOH) as the reference electrode. A total of 2 M NaOH + 0.1 M NaBH4 was on the anode side, and 2.0 M HCl + 4.5 M H2O2 was on the cathode side. High purity nitrogen gas was bubbled in the anolyte for 30 min before tests to eliminate O2 and CO2. The cell testing was performed using American Princeton VMPIII constant potential rectifier, and the data were analyzed by EC-Lab.

The schematic illustration of the direct NaBH4/H2O2 fuel cell. 1 Anode electrode; 2 cathode electrode; 3 reference electrode; 4 0.5 M NaBH4 + 2 M NaOH; 5 4.5 M H2O2 + 2.0 M HCl; 6 the activated Nafion 212 membrane
Results and discussion
Physical characterization of the carbon-supported electrocatalysts
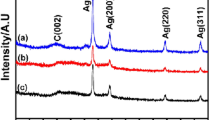
The XRD spectra of different carbon catalysts are shown in Fig. 2. There are four diffraction peaks positioned at 2θ = 38.3°, 44.3°, 64.3°, and 77.3° on the spectra of Ag/C and can be indexed to the Ag(111), Ag(200), Ag(220), and Ag(311) planes, indicating the fcc structure of Ag. From Fig. 2, we can see clearly that the peaks on spectra of Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C match well with those on Ag/C, and the peak value becomes higher with Ag content increasing. No diffraction peaks of Ni or its oxides are observed for Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C.

XRD spectra of different carbon catalysts
The mean diameters of Ag/C and Ni@Ag/C catalysts can be estimated from the Scherrer equation using the diffraction peak of Ag(111):
where D is the average crystal size (nm), λ is the X-ray wavelength (1.54056 Å for Cu Kɑ radiation), B is the full width at half-maximum in radians (FWHM), and θ is the Bragg angle (rad). The calculated mean crystal sizes of Ag/C, Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C were 17.1, 15.1, 13.2, 12.9, and 12.7 nm, respectively.
Figure 3a, b is typical TEM and HRTEM images of the Ni1@Ag1/C nanoparticles. It could be clearly seen that most of the metal nanoparticles are roughly spherical in shape; agglomerates with different sizes are visible. Figure 3c shows the histogram of the diameter distribution of the nanoparticles. From the histogram, the average diameter of the nanopaticles was estimated to be 14 nm. The measured particle size was consistent with the XRD results. EDS was used to determine the chemical composition of the Ni@Ag/C catalysts, and results showed that the atomic ratios for Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C are 39:61, 51:49, 42:58, and 67:33, respectively. These ratios are in good agreement with the original inputs.

a, b TEM images of Ni1@Ag1/C nanoparticles, c paticle size distribution carried out on TEM image
Cyclic voltammetry
The CVs recorded for the Ag/C, Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, Ni2@Ag1/C, and Ni/C electrodes with 2 M NaOH and 2 M NaOH + 0.1 M NaBH4 solution at a scan rate of 50 mV s−1 in the potential range of −0.9 to 0.7 V (vs MOE) are shown in Fig. 4. As shown in Fig. 4, the shapes of the voltammograms of borohydride oxidation on Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C electrodes were similar to those of the Ag/C electrode. In absence of NaBH4, a pair of redox peaks b1 (about 0.4 V) and b5 (0∼0.1 V) in Fig. 4a was assigned to Ag/Ag2O redox couple in alkaline media [10], which coincided with that in Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C. Another peak b3 in Fig. 4a that also could be seen in Fig. 4b–d could be attributed to the reduction of higher Ag oxides [38]. In Fig. 4b–f, a pair of redox peaks b2 and b4 appearing at 0.5 and 0.3 V have also been observed in alkaline solution, which correspond to the oxidation-reduction processes of nickel hydroxide [24, 35]. The occurrence of nickel hydroxide indicated that the Ni nanoparticles were not wrapped up by Ag completely. In the presence of NaBH4, peak a1 in Fig. 4a was the oxidation peak of BH ‐4 , and the current still increased with the formation of Ag2O. So, the direct oxidation of BH ‐4 mainly took place upon multilayered Ag2O, which coincided with that in Fig. 4b–e. The mechanism could be expressed as following [39]:

Voltammograms of different anode catalysts in 2 M NaOH and 2 M NaOH + 0.1 M NaBH4: a Ag/C, b Ni1@Ag1.5/C, c Ni1@Ag1/C, d Ni1.5@Ag1/C, e Ni2@Ag1/C, f Ni/C
Obviously, there was another peak a2 (about 0.5 V) in the positive potential sweep on the Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C, which coincides with peak b2. The reason might be that the nanoparticle of a core-shell structure had dual physicochemical performance as core metal Ni and as coating shell metal Ag.
In the reverse potential sweep, peaks c1 and c3 coincide with the peaks b3 and b5, which were the reduction peaks of Ag oxides.
The onset oxidation potentials and peak current densities of BH ‐4 for Ag/C, Ni@Ag/C, and Ni/C catalysts are summarized in Table 1. According to Table 1, the onset potentials of BH ‐4 for Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C were more negative than Ag/C and Ni/C catalyst, suggesting the improved catalytic activity of the core-shell nanoparticles. Although the content of Ag in Ni@Ag/C catalysts was less than that in Ag/C, the peak current density at Ni@Ag/C catalysts did not reduce. Among the six electrodes, the peak current density on the Ni1@Ag1/C was the highest.
Chronoamperometry
Figure 5 shows the chronoamperograms of different carbon catalysts at −0.2 V in 2 M NaOH + 0.1 M NaBH4. As shown in Fig. 5, a decrease in the current with time was found in each catalyst electrode. After the application of the set potential for 30 s, the current tended to be stable. The current values of Ni@Ag/C electrode were higher than those of Ag/C. This result indicated that using Ni as the core metal can greatly improve the catalytic activity for the BH ‐4 electrooxidation. The order of electrocatalytic activity was Ni1@Ag1/C > Ni1.5@Ag1/C > Ni2@Ag1/C > Ni1@Ag1.5/C > Ag/C. The current densities were 15.8, 10.1, 6.8, 5.2, and 2.2 mA cm−2, respectively. It was clear that Ni1@Ag1/C would exhibit the best catalytic performance among all the four electrodes.

Chronoamperometry of different carbon catalysts at −0.2 V in 2 M NaOH + 0.1 M NaBH4
Kinetic parameters for BH ‐4 electrocatalytic oxidation reaction
With the aim to determine the kinetic parameters and the total number of the electrons, the linear scan voltammetry with rotating disk electrode (LSV RDE) was performed. Figure 6 shows the results of RDE LSVs of the five electrodes recorded in 0.1 M NaBH4 + 2 M NaOH solution at 0.003-V s−1 scan rate and using electrode rotation speed (ω, rpm) in 1400 rpm at 25 °C. At high potentials, the borohydride is depleted inside the porous electrode structure and is not replenished by the rotation effects, thus leading to a bell-shaped curve [40].

Linear scan voltammograms with rotating disk electrode (RDE LSVs) of the studied electrocatalysts recorded in 0.1 M NaBH4 + 2 M NaOH at the electrode rotation speed of 1400 rpm
The data from LSVs recorded using different rotation speeds were used to determine the kinetic parameters of the BOR at the investigated electrocatalysts. The number of electrons exchanged, n, was calculated using the Koutecky–Levich equation (Eqs. (9–10)), which defines the overall current density dependence on the value of diffusion-limited current density (i l) and kinetic current density (i k) [41], where i is the disk current density (A), n the total number of electrons exchanged, F the Faraday constant (96,485 C mol−1), k the apparent rate constant (cm s−1), C *O the concentration of the borohydride (mol cm−3), A the area of the electrode, D the diffusion coefficient of the electrochemically active species (1.6 × 10−5 cm2 s−1) [4], ω the angular rotation rate of the electrode (radians s−1), and ν is the kinematic viscosity of the solution (1.19 × 10−2 cm2 s−1). According to Eq. (10), the plot of the reciprocal value of the disk current density i −1 versus ω −1/2 should be linear, and Fig. 7 shows the results of the five electrodes obtained in 0.1 M NaBH4 + 2 M NaOH solution at 0.003 V s−1 scan rate and using electrode rotation speed (ω, rpm) in the range from 200 to 2600 rpm. The slopes of the i −1 versus ω −1/2 plot were used to calculate n, and Table 2 summarizes the results.

Plots of i −1 (cm2 mA−1) vs ω −1/2 (radians s−1) of the studied electrocatalysts for 0.1 M NaBH4 + 2 M NaOH solution using electrode rotation speed (ω, rpm) in the range from 200 to 2600 rpm
It was noticed that the n values transferred on Ag/C was lower than that measured in Ref [4, 10, 11]. The reason might be that the ratio of [OH−]/[BH ‐4 ] was a little low and leading to negligible BH3OH− and H2 production. The adsorbed intermediates might block the electrode surface for further oxidation of borohydride. Ag/C nanoparticles do not show any significant activity toward H2 oxidation. So, the higher n values reported for Ni@Ag/C than for Ag/C might originate from the trapping of the BOR intermediates in the active layer, increasing their “residence time” and improving their subsequent oxidation [11]. According to Table 2, the n value of Ni@Ag/C is close to that calculated for the other Ag-based in the literature. The n value of Ni1@Ag1/C was higher than that of the other three catalysts, indicating better BOR performance than the other three catalysts.
Figure 8 shows the Tafel plots for Ag/C and Ni@Ag/C catalysts generated from points of the Tafel region of the corresponding LSV data at 1400 rpm. The mass-transfer-corrected (kinetic) current density i k was calculated from the measured i (total) value by the standard 1/i (total) = 1/i k +1/ i l (Eq. (9)). The Tafel slopes b and exchange current density i 0 were also listed in Table 2. We can observe in Table 2 that the values of i 0 are negligibly small and thus considered meaningless in evaluating the electrocatalytic activity of anode catalysts [42], and the i 0 of the five anodes was rather smaller than some recently reported results in the literatures [4, 11, 24, 40]. All of the b values were close to 0.12 V dec−1, and some were a little higher than 0.12 V dec−1 when based solely on electrode kinetic considerations. Higher Tafel slopes may be attributed to mixed control scenarios, such as electrode kinetics combined with mass transfer and/or ionic conductivity effects in the catalyst layer [43, 44]. According to Table 2, the Tafel slopes of Ni@Ag/C were smaller than those of Ag/C. Since a lower value of Tafel slope is desirable, high Tafel slope values indicate inferior BOR activities. Moreover, the exchange current densities calculated for Ni@Ag/C are higher than those for Ag/C. These data indicated that the Ni@Ag/C catalysts reduced the influence of the electrode potential upon the activation energy of BH ‐4 oxidation reaction and made the electrode reaction much easier [45]. The Ni1@Ag1/C catalyst exhibited relatively smaller b value.

Tafel plots for Ag/C and Ni@Ag/C catalysts generated from the corresponding LSV data at 1400 rpm
LSVs RDE data were also used for evaluation of apparent activation energy (E appa ) for the BOR on Ag/C and Ni@Ag/C electrocatalysts in 0.1 M NaBH4 + 2 M NaOH. Data of the kinetic current density in the temperature region 25–55 °C at 1400 rpm shown in Fig. 9 was used for application of the Arrhenius equation (Eq. (11)),

Arrhenius plot for borohydride oxidation on Ag/C and Ni@Ag/C electrocatalysts
assuming that E appa does not depend on temperature and i k does not depend on potential [46]. The E appa values of the different electrocatalysts were listed in Table 2; this value of E appa is comparable to the values reported on different electrocatalysts, such as Ag/C Pt/C, Pt0.75Co0.25, and Pt0.75Ni0.25 (35.0, 25.3, and 20.2 kJ mol−1, respectively) [27, 47]. It was clear seen that Ni1@Ag1/C had the lowest E appa among the five catalysts. This value was significantly lower than that reported for BOR at Au electrode [19, 20]. This phenomenon further demonstrated that Ni@Ag/C could apparently improve the electrode kinetics for BH ‐4 oxidation, and it may exhibit better BH ‐4 electrooxidation kinetics than the other catalysts.
The reaction order of the BOR at the investigated electrocatalysts was determined from LSV data for different NaBH4 concentrations ranging from 0.04 to 0.16 M using Eq. (12) [19]:
where z is the constant and β is the order of the reaction with respect to BH ‐4 . Figure 10 shows the relationship between the kinetic current density and concentrations of borohydride. Therefore, the value of β can be determined from the slope of log i vs log C plot (Table 2).

Relationship between the kinetic current density and concentrations of borohydride and hydroxide. The data were collected at −0.1 V. Other conditions as shown in Fig. 6
Cell performance
Figure 11 shows the cell polarization and power density curves for DBHFC using Ag/C, Ni1@Ag1.5/C, Ni1@Ag1/C, Ni1.5@Ag1/C, and Ni2@Ag1/C as anode catalysts. The anolyte was 2 M NaOH + 0.1 M NaBH4 solution, and the catholyte was 2 M HCl + 4.5 M H2O2 solution. Main performance parameters of the DBHFC are summarized in Table 3. According to Table 3, the open circuit voltages (OCVs) of the cell using Ni@Ag/C as the anode catalysts are higher than those of Ag/C. The reason could be that with the introduction of Ni core, the performance of the core-shell catalysts was improved compared with Ag catalyst. Obviously, all the OCVs were lower than the theoretical value of DBHFC (3.01 V). The low value was probably caused by mixed potential from at the anode and the cathode [48]. As shown in Fig. 11, the cell voltages of DBHFC decreased with the increasing of current density. The DBHFC with Ni@Ag/C anode had smaller polarization for BH ‐4 oxidation than the one with Ag/C anode for the reason that Ni@Ag/C core-shell could improve the performance of DBHFC. It was found that DBHFC with Ni1@Ag1/C as anode catalyst gave the best performance. The maximum power density is 8.54 mW cm−2 at a current density of 8.42 mA cm−2.

Cell polarization and power density curves of the DBHFC using various carbon anode catalysts
All the research results show that the core-shell catalyst has higher catalytic activity than Ag/C catalyst. It is well known that the presence of another metal in the parent lattice affects both the electronic nature and the crystal structure, which in turn influence the catalytic behavior of bimetallic nanoparticles. Both electronic structure and surface atoms involved in the catalytic performance. The linear relationship between d-band center energy and chemisorption energy was established [49]. C.A. Kuhnen [50] used the density of states (DOS) to analyze the electronic properties of Ni/Ag bilayers. The results showed that there existed a strong interaction between s, p, and d states of Ni with d states of Ag and the charge could transfer from Ag to Ni sites, since in this manner, the electrons leave the high energy occupied states at Ag to occupy the empty d down states at relative low energies at Ni sites. This electron transformation may lead to the increase in vacancies at the Ag 4d-obital. The higher-lying d states of the Ag atoms lowered its average energy and then reveal the moderate adsorption capacity of BH ‐4 , which improves the catalytic activity.
Conclusion
In the present study, carbon-supported Ni@Ag core-shell nanoparticles were successfully synthesized by impregnation reduction method in aqueous solution. The average particle size was approximately 13 nm. The Ni@Ag/C catalysts represented higher electrochemical catalytic activity for BH ‐4 electrooxidation than Ag/C catalyst in the alkaline solution for the reason that core-shell nanoparticles with some shell thickness could significantly improve the electrocatalytic activity of the catalyst. Among the as-prepared catalysts, Ni1@Ag1/C revealed the highest electrocatalytic activity to the direct oxidation of BH ‐4 as evidenced by higher current densities along with higher number of electrons transferred during BOR and lower activation energy. The DBHFC employing Ni1@Ag1/C as anode catalyst and Pt mesh (1 cm2) as cathode electrode obtained the maximum anodic power density as high as 8.54 mW cm−2 at a discharge current density of 8.42 mA cm−2 at 25 °C.
Therefore, the carbon-supported Ni@Ag nanoparticles with high performance and low cost are expected to be a promising anode catalyst for the application of DBFC.
References
Feng RX, Dong H, Wang YD, Ai XP, Cao YL, Yang HX (2005) A simple and high efficient direct borohydride fuel cell with MnO2-catalyzed cathode. Electrochem Commun 7:449–452
Peled E, Livshits V, Duvdevani T (2002) High-power direct ethylene glycol fuel cell (DEGFC) based on nanoporous proton-conducting membrane (NP-PCM). J Power Sources 106:245–248
Ma J, Sahai Y, Buchheit RG (2010) Direct borohydride fuel cell using Ni-based composed anodes. J Power Sources 195:4709–4713
Chatenet M, Micoud F, Roche I, Chainet E (2006) Kinetics of sodium borohydride direct oxidation oxygen reduction sodium hydroxide electrolyte. Part I: BH4 − electro-oxidation on Au and Ag catalyst. Electrochim Acta 51:5459–5467
Cao D, Gao Y, Wang G, Miao R, Liu Y (2010) A direct NaBH4-H2O2 fuel cell using Ni foam supported Au nanoparticles as electrodes. Int J Hydrogen Energy 35:807–813
Liu BH, Li ZP, Suda S (2004) Electrocatalysts for the anodic oxidation of borohydrides. Electrochim Acta 49:3097–3105
Cao D, Chen D, Lan J, Wang G (2009) An alkaline direct NaBH4-H2O2 fuel cell with high power density. J Power Sources 190:346–350
Gardiner JA, Collat JW (1965) Kinetics of the stepwise hydrolysis of tetrahydroborate ion. J Am Chem Soc 10:1692–1700
Kim JH, Kim HS, Kang YM, Song MS, Rajendran S, Han SC, Jung DH, Lee JY (2004) Carbon-supported and unsupported Pt anodes for direct borohydride liquid fuel cells. J Electrochem Soc 151:A1039–A1043
Concha BM, Chatenet M (2009) Direct oxidation of sodium borohydride on Pt, Ag and alloyed Pt-Ag electrodes in basic media.PartI: Bulk electrodes. Electrochim Acta 54:6119–6229
Concha BM, Chatenet M (2009) Direct oxidation of sodium borohydride on Pt, Ag and alloyed Pt-Ag electrodes in basic media.PartII: Carbon-supported nanopaticles. Electrochim Acta 54:6130–6139
Cenk C, San FGB, Sarac HI (2008) Effects of operation conditions on direct borohydride fuel cell performance. J Power Sources 185:197–201
Yang JQ, Liu BH, Wu S (2009) Carbon-supported Pd catalysts: Influences of nanostructure on their catalytic performances for borohydride electrochemical oxidation. J Power Sources 194:824–829
Liu BH, Li ZP, Suda S (2003) Anodic oxidation of alkali borohydrides catalyzed by nickel. J Electrochem Soc 150:A398–A402
Wang KL, Lu JT, Zhuang L (2007) A current decomposition study of the borohydride oxidation reaction at Ni electrodes. J Phys Chem C 111:7456–7462
Duan DH, Liu SB, Sun YP (2012) Analysis of the kinetics of borohydride oxidation in Cu anode for direct borohydride fuel cell. J Power Sources 210:198–203
Zhang D, Ye K, Cao D, Wang B, Cheng K, Li Y, Wang G, Xu Y (2015) Co@MWNTs-Plastic: A novel electrode for NaBH4 oxidation. Electrochim Acta 156:102–107
Zhang D, Ye K, Cheng K, Cao D, Yin J, Xu Y, Wang G (2014) High electrocatalytic activity of cobalt–multiwalled carbon nanotubes–cosmetic cotton nanostructures for sodium borohydride electrooxidation. Int J Hydrogen Energy 39:9651–9657
Santos DMF, Sequeira CAC (2010) Cyclic voltammetry investigation of borohydride oxidation at a gold electrode. Electrochim Acta 55:6775–6781
Cheng H, Scott K (2006) Determination of kinetic parameters for borohydride oxidation on a rotating Au disk electrode. Electrochim Acta 51:3429–3433
Sanli E, Celikkan H, Uysal BZ, Aksu ML (2006) Anodic behavior of Ag metal electrode in direct borohydride fuel cells. Int J Hydrogen Energy 31:1920–924
Liu BH, Li ZP (2009) A review: hydrogen generation from borohydride hydrolysis reaction. J Power Sources 187:527–534
Hosseini MG, Abdolmaleki M (2013) Synthesis and characterization of porous nanostructured Ni/PdNi electrode towards electrooxidation of borohydride. Int J Hydrogen Energy 38:5449–5456
He PY, Wang Y, Wang XY, Pei F, Wang H, Liu L, Yi LH (2011) Investigation of carbon supported Au-Ni bimetallic nanoparticles as electrocatalyst for direct borohydride fuel cell. J Power Sources 196:1042–1047
Geng XY, Zhang HM, Yea W, Ma YW, Zhong HX (2008) Ni-Pt/C as anode electrocatalyst for a direct borohydride fuel cell. J Power Sources 185:627–632
Wang GJ, Gao YZ, Wang ZB, Du CY, Wang JJ, Yin GP (2010) Investigation of PtNi/C anode electrocatalysts for direct borohydride fuel cell. J Power Sources 195:185–189
Šljukić B, Milikić J, Santos DMF, Sequeira CAC (2013) Carbon-supported Pt0.75M0.25 (M= Ni or Co) electrocatalysts for borohydride oxidation. Electrochim Acta 107:577–583
Feng RX, Dong H, Cao YL, Ai XP, Yang HX (2007) Agni-catalyzed anode for direct borohydride fuel cells. Int J Hydrogen Energy 32:4544–4549
Goodman DW (1990) Chemistry on monolayer metallic films. Ultramicroscopy 34:1–9
Chen DH, Wang SR (2006) Protective agent-free synthesis of Ni-Ag core-shell nanoparticles. Mater Chem Phys 100:468–471
Yuan QB, Duan DH, Ma YH, Wei GQ, Zhang ZL, Hao XG, Liu SB (2014) Performance of nano-nickel core wrapped with Pt crystalline thin film for methanol electro-oxidation. J Power Sources 245:886–891
Lin R, Cao CH, Zhao TT, Huang Z, Li B, Wieckowski A, Ma JX (2013) Synthesis and application of core–shell Co@Pt/C electrocatalysts for proton exchange membrane fuel cells. J Power Sources 223:190–198
Zhou W, Lee JY (2007) Highly active core–shell Au@Pd catalyst for formic acid electrooxidation. Electrochem Commun 9:1725–1729
Sarkar A, Manthiram A (2010) Synthesis of Pt@Cu core-shell nanoparticles by galvanic displacement of Cu by Pt4 + ions and their application as electrocatalysts for oxygen reduction reaction in fuel cells. J Phys Chem C 114:4725–4732
Duan DH, Liang JW, Liu HH, You X, Wei HK, Wei GQ, Liu SB (2015) The effective carbon supported core–shell structure of Ni@Au catalysts for electro-oxidation of borohydride. Int J Hydrogen Energy 40:488–500
Duan D, Liu H, You X, Wei H, Liu S (2015) Anodic behavior of carbon supported Cu@Ag core-shell nanocatalysts in direct borohydride fuel cells. J Power Sources 293:292–300
Baletto F, Mottet C, Rapallo A, Rossi G, Ferrando R (2004) Growth and energetic stability of AgNi core-shell clusters. Surf Sci 566–568:192–196
Burstein GT, Newman RC (1980) Anodic behavior of scratched silver electrodes in alkaline media. Electrochim Acta 25:1009–1014
Sanli E, Uysal BZ, Aksu ML (2008) The oxidation of NaBH4 on electrochemically treated silver electrodes. Int J Hydrogen Energy 33:2098–2099
Atwan MH, Northwood DO, Gyenge EL (2007) Evaluation of colloidal Ag and Ag-alloys as anode electrocatalysts for direct borohydride fuel cells. Int J Hydrogen Energy 32:3116–3125
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications, 2nd edn. John Wiley and Sons, New York, pp 337–341
Lai Y, Li Y, Jiang L, Xu W, Lv X, Li J, Liu Y (2012) Electrochemical behaviors of co-deposited Pb/Pb-MnO2 composite anode in sulfuric acid solution - Tafel and EIS investigations. J Electroanal Chem 671:16–23
Gyenge EL (2005) Dimensionless numbers and correlating equations for the analysis of the membrane-gas diffusion electrode assembly in polymer electrolyte fuel cells. J Power Sources 152:105–121
Mirkin MV, Yang H, Bard AJ (1992) Borohydride oxidation at a gold electrode. J Electrochem Soc 139:2212–2217
Burstein GT (2005) A hundred years of Tafel’s Equation: 1905–2005. Corros Sci 47:2858–2870
Liu DM, Huang WJ, Si TZ, Zhang QA (2013) Hydrogen storage properties of LiBH4 destabilized by SrH2. J Alloys Compds 551:8–11
Ivan S, Jelena K, Jadranka M, Biljana S, Zorica K-P, Slavko M, Scepan M (2016) Radiolitically synthesized nano Ag/C catalysts for oxygen reduction and borohydride oxidation reactions in alkaline media, for potential applications in fuel cells. Energy 101:79–90
de León CP, Walsh FC, Patrissi CJ, Medeiros MG, Bessette RR, Reeve RW, Lakeman JB, Rose A, Browning D (2008) A direct borohydride–peroxide fuel cell using a Pd/Ir alloy coated microfibrous carbon cathode. Electrochem Commun 10:1610–1613
Long NV, Hien TD, Asaka T, Ohtaki M, Nogami M (2011) Synthesis and characterization of Pt-Pd alloy and core-shell bimetallic nanoparticles for direct methanol fuel cells (DMFCs): Enhanced electrocatalytic properties of well-shaped core-shell morphologies and nanostructures. Int J Hydrogen Energy 36:8478–8491
Kuhnen CA (1996) Magnetic and electronic structure of Ni/Ag bilayers. Solid State Commun 98:123–128
Acknowledgments
This work was supported by a grant from the Natural Science Foundation of Shanxi Province (No. 2015011028).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Duan, D., Wang, Q., Liu, H. et al. Investigation of carbon-supported Ni@Ag core-shell nanoparticles as electrocatalyst for electrooxidation of sodium borohydride. J Solid State Electrochem 20, 2699–2711 (2016). https://doi.org/10.1007/s10008-016-3285-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3285-5