
Abstract
In this paper, we report the successful synthesis of multiwalled carbon nanotubes supported by gold-cobalt (AuxCo100 − x/MWCNT) nanoparticles to develop a novel electrocatalyst for anodic application in direct borohydride fuel cells. X-ray diffraction spectroscopy, X-ray photoelectron spectroscopy, field emission scanning electron microscopy and transmission electron microscopy are employed to examine the crystalline structure, chemical composition and morphology of the prepared electrocatalysts. Cyclic voltammetry, electrochemical impedance spectroscopy and chronoamperometry tests are used for electrocatalytic characterizations of developed electrocatalysts. Using AuxCo100 − x/MWCNT electrocatalyst, the fundamental kinetics parameters of electrocatalytic performance (current density, exchanged electrons number and apparent activation energy) for borohydride electrooxidation are investigated. Results reveal that among all bimetallic electrocatalysts, the Au74Co26/MWCNT electrocatalyst exhibits the highest specific activity (24.15 mA.cm− 2) and Au49Co51/MWCNT shows the highest mass activity (1127.03 mA.mg− 1) for \({BH}_{4}^{-}\) electrooxidation. The lowest apparent activation energy (8.22 kJmol− 1) and smallest charge transfer resistance (134.9 Ω) suggest the best electrocatalytic activity of Au74Co26/MWCNT electrocatalyst toward borohydride oxidation. The exchanged electron number for the Au74Co26/MWCNT electrocatalyst for borohydride electrooxidation at \(303 \text{K}\)is estimated as 4.70.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, developing alternative energy sources is one of the most vital issues. The fuel cell is an alternative energy source that converts chemical energy directly to electrical energy. Less pollution, higher efficiency, simple structure, instant recharging, easy storage and less noise make fuel cells as a potential energy source [1,2,3,4]. Researchers concentrated on direct borohydride fuel cells (DBFC) due to some limitations in alcohol fuel cells, such as low electrochemical activity, CO2 generation, voltage loss, significant anodic polarization and relatively high corrosion activity [5]. Sodium borohydride (NaBH4) was first confirmed as a fuel for fuel cells in 1960 [6]. Since then, NaBH4 has been considered to be an efficient hydrogen source for fuel cells due to its high hydrogen content (10.6%), a higher theoretical open circuit voltage of 1.64 V than other fuels, high energy density, high power density, significant chemical stability in alkaline medium, non-toxicity and it is trouble-free to store and transport [7, 8]. In an alkaline solution, the complete electrooxidation of borohydride anion (BH4−) generates eight electrons [9] at the anode and oxygen reduction at the cathode and produces energy, shown as Eq. (1)
In addition, the number of generated electrons in the electrooxidation of Ethanol [10], Methanol [11], Hydrazine [12] and Hydrogen [13] is lower than that of NaBH4. A suitable electrocatalyst is a crucial factor for enhancing the energy production performance of DBFCs. Many researchers have reported different types of monometallic and bimetallic electrocatalysts to enhance the electrocatalytic performances of DBFCs. Due to decomposition reactions, the composition of electrocatalysts with selectivity and activity has an important influence on electrooxidation reactions. Decomposition reactions in DBFC result in energy loss and decrease the efficiency of the cell [14]. So, catalytic selectivity should be considered to develop the most suitable electrocatalyst. The anodic catalyst is one of the important parameters to optimize its catalytic performance in the electrochemical oxidation of BH−4. As per a previous study, Au shows good catalytic performance in borohydride oxidation [15, 16]. However, recent studies have claimed that Au cannot promote direct borohydride electrooxidation completely, so Au alone is challenging to use as an efficient anode electrocatalyst. Thus, developing bimetallic catalysts containing Au can be a good strategy. Many researchers revealed that bimetallic electrocatalysts show better electrocatalytic performance than monometallic electrocatalysts due to the synergic effects of bimetallic catalytic materials. The expansive cost and slow electrooxidation kinetics of borohydride ions on monometallic Au anode catalyst limit the application of Au in DBFC [17, 18]. Noble metals [Au (gold), Pt (platinum), Pd (Palladium), Os (Osmium), Ag (Silver)] with the 3d transition metals [Ni (nickel), Co (Cobalt), Zn (Zinc), Fe (Iron), Cu (Copper)] and their alloys such as Au-Ni, Au-Co, Au-Fe, Au-Zn, Au-Cu, etc. show better electrocatalytic activity and lower cost than monometallic Au electrocatalysts [19]. Hence, enhancement of electrocatalytic performance of Au and Co electrodes is important for advancing the performance and lessening the cost of DBFCs. Taek Hyun Oh et al. [20] confirmed that the performance of the electrocatalyst with multiwalled carbon nanotubes (MWCNTs) is better than that of the catalyst with XC-72 owing to the good electrical conductivity of MWCNTs. Duan et al. [7] have reported that carbon-supported Co-Au bimetallic electrocatalysts exhibit higher electrocatalytic activity and stability than monometallic Au/C catalysts for BH4− electrooxidation.
In this study, AuxCo100 − x/MWCNT electrocatalysts are synthesized with 20 wt% metal loadings by a simple chemical reduction method for borohydride oxidation. X-ray diffraction spectroscopy (XRD), energy dispersive X-ray spectroscopy (EDX), X-ray photoelectron spectroscopy (XPS), field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) are employed as physicochemical characterization techniques to examine the crystalline structure, chemical composition and morphology of the prepared electrocatalysts. The electrocatalytic properties of the prepared catalysts are tested by cyclic voltammetry (CV). The charge transfer kinetics is investigated by electrochemical impedance spectroscopy (EIS). A chronoamperometry (CA) study examines the catalysts’ durability. All the electrochemical tests confirm the enhanced electrocatalytic performance of the bimetallic system.
2 Experimental
2.1 Chemicals and reagents
Chloroauric acid extra pure [HAuCl4] (MW-339.79 g/mol) was purchased from LOBA CHEMIE PVT.LTD. Cobalt(III) nitrate hexahydrate purified [CO(NO3)2.6H2O] (MW-291.04 g/mol), Sodium borohydride [NaBH4] (MW-37.83 g/mol), tri-sodium citrate dihydrate [C6H5Na3O7.2H2O] (MW-294.1 g/mol), sodium hydroxide [NaOH] (MW-40.00 g/mol) pellets, nitric acid [HNO3] and hydrochloric acid [HCl] were purchased from MERCK, India. MWCNT was purchased from Nanocyl 3100™. Triple-distilled water was used throughout the experiment.
2.2 Synthesis of bimetallic Aux-Co100−x/MWCNT catalysts
At first, we functionalized the MWCNT by acid treatment. 250 mg MWCNT was dispersed into 40 ml aqua regia using the ultrasonic bath for 30 min. After that, the mixture was stirred under continuous magnetic stirring overnight at 200 rpm and \(333 K\) temperature. The acidulated MWCNT was washed with deionized water and ethanol several times until a neutral pH was obtained. Then the mixture was dried at 253\(K\) for 12 h and we collected the functionalized MWCNT.
First, 46.4 mg of MWCNT was dispersed homogeneously into 30 ml of deionized water and sonicated for 30 min. After that, the mixture was agitated for half an hour using a magnetic stirrer. A continuous magnetic stirring process is adopted here. 20 ml aqueous solution of 20 mg of HAuCl4 was added with the previous mixture. After 30 min, 10 ml aqueous solution of 10 mg sodium citrate was mixed with the solution. 15 min later, 10 ml aqueous solution of NaBH4 (20 mg) was dropped gently into the solution. Then, the final mixture was stirred under continuous magnetic stirring for 5 h. After that, the mixture was washed with deionized water several times until obtained a neutral pH. Finally, the precipitate was dried for 8 h at \(253 K\) and marked as S-1 (Au100/MWCNT). Next, MWCNT (31 mg) was dispersed homogeneously into 30 ml of deionized water and sonicated for 30 min. After that, the mixture was stirred for 30 min using a magnetic stirrer. A continuous magnetic stirring process is adopted here. 10 ml aqueous solution of 10 mg of cobalt(II) nitrate hexahydrate was added with the previous mixture. After 30 min, 10 ml aqueous solution of 10 mg sodium citrate was mixed with the solution. 15 min later, 5 ml aqueous solution of NaBH4 (10 mg) was dropped gently into the solution. Subsequently, a 10 ml aqueous solution of 10 mg HAuCl4 was added. After 15 min, 5 ml aqueous solution of NaBH4 (10 mg) was added dropwise. Then, the final mixture was stirred under continuous magnetic stirring for 5 h. After that, the mixture was washed with deionized water several times until obtained a neutral pH. Finally, the precipitate was dried for 8 h at \(253 \text{K}\) and marked as S-2 (Au74Co26/MWCNT). Other catalysts, S-3 (Au59Co41/MWCNT) and S-4 (Au49Co51/MWCNT) were synthesized similarly by varying the amount of metal precursors. The metal loading in each sample was maintained at 20 wt%. The information about the Au and Co weight ratio of the developed electrocatalysts is given in Table 1.
2.3 Physical characterization of electrocatalysts
The X-ray diffraction (XRD) spectra of four different electrocatalysts (S-1, S-2, S-3 & S-4) were recorded by a Bruker D8 Advance X-ray diffractometer with CuKα radiation (λ= 1.54 Å) in 2θ angular regions between 20° and 80° with step size 0.02°. The average crystallite sizes for the synthesized electrocatalysts were calculated according to the full-width half maxima (FWHM). The scanning electron microscope (ZEISS Gemini SEM) and tunneling electron microscopy (JEOL, 2100 F) were used for the morphological analysis of the electrocatalysts. The chemical compositions of the prepared electrocatalysts were examined by an energy-dispersive X-ray spectroscopy (EDX) system. The bonding, compositions and surface chemical state of the synthesized electrocatalysts were analyzed by XPS using PHI 5000 Versa Probe (II), FEI Inc- ray electron spectrometer.
2.4 Electrochemical characterization of electrocatalysts
All the electrochemical investigations of as-prepared AuxCo100 − x/MWCNT electrocatalysts were performed by a potentiostat/galvanostat (PGSTAT302N, Autolab, Metrohm Electrochemistry Workstation). A 3 mol KCl saturated Ag/AgCl electrode was employed as a reference electrode and a mesh of platinum wire with dimensions of 1 cm × 1 cm (50 meshes) was used as the counter electrode. In this study, we used a glassy carbon electrode (GCE) with having 5 mm diameter as a working electrode. Prior to each experiment, the GCE was thoroughly cleansed with distilled water and ethanol. The catalyst ink was made using Nafion (Nafion™ NR 50, Merck) as a binder. After complete dissolution of 50 mg Nafion in 50 ml of ethanol, 8 mg of AuxCo100−x/MWCNT nanopowder was added to the 1 ml of Nafion solution. A homogenous catalyst ink was created by sonicating the mixture for 25 min in an ultrasonic bath. We dropped cast 5 µl of catalyst ink onto the GCE surface and dried it under a 100 W incandescent light for 1.5 h. The geometric area of the GCE surface was used to compute the current densities. A single-compartment borosilicate beaker was utilized as the electrolyte cell, and triple distilled water was used to prepare the electrolyte solutions (0.5 M NaOH and 0.5 M NaOH + 0.01 M NaBH4). To quantify the electrocatalytic removal of O2 and CO2 from the electrolyte solutions, pure N2 gas was bubbled in the mixtures for 25 min.
3 Results and discussion
3.1 Physical characterization of the MWCNT-supported electrocatalysts
The XRD diffractograms of different electrocatalysts are shown in Fig. 1. In all catalysts, the first broad diffraction peak was visible at 2θ = 25.83◦. The peak was connected to the hexagonal graphite structures (002) in MWCNT (JCPDS No. 75-1621). The remaining five strong diffraction peaks which are located at around 2θ = 38.4°, 44.53°, 64.87°, 77.84°, 82.01° and corresponding face-centered cubic (fcc) Au’s (111), (200), (220), (311) and (222) planes respectively were well matched with the typical Au peaks (JCPDS No. 04-0784) [21]. The major peaks of AuxCo100−x/MWCNT were consistent with those of Au/MWCNT, although neither Co nor its oxide showed any diffraction peaks. It is most likely because only the Au atom crystallises to form a crystal in the Au-Co nanoparticles, whereas Co exists in the form of amorphous Co atoms adsorbed on the gold crystal [7, 22]. The EDS results have verified the existence of Co.

Microstructure of S-1, S-2, S-3, and S-4 anode electrocatalysts
The Debye-Scherrer equation was used to determine the electrocatalyst’s average crystal size.
where D is the average crystallite size (nm), K is known as Scherrer’s constant (K = 0.94), λ is the wavelength of X-ray (1.54 Å for Cu Kα radiation), β is the full width at half maxima (FWHM) in radians and θ is the Bragg angle in radians. The evaluated crystallite size of S-1, S-2, S-3 and S-4 samples were 10.44, 4.09, 7.12 and 6.34 nm, respectively.
The synthesized electrocatalysts are depicted in typical FESEM images in Fig. 2. All electrocatalysts have an aggregation of metal particles with varying sizes and a generally sphere-like form. On MWCNT, the particles are evenly spread. To investigate the chemical composition of AuxCo100−x/MWCNT electrocatalysts, energy-dispersive spectroscopy (EDS) was done. Figure 3 shows the EDS images of all prepared electrocatalysts. All the EDS spectra showed the characteristic peaks for C, O, Au and Co.

FESEM images of S-1, S-2, S-3 and S-4 bimetallic electrocatalysts

EDS images of S-1, S-2, S-3, and S-4 electrocatalysts
Figure 4a–c show TEM, HRTEM and SAED images of the S-2 electrocatalyst. The TEM image (Fig. 4a) reflects the nearly sphere-like nanoparticles are well distributed in MWCNT. The HR-TEM image (Fig. 4b) approves the presence of monodispersed Au nanoparticles of around 5 nm in mean diameter as reflected. From the HR-TEM analysis, it is visible that nanoparticles with an interplanar spacing of 0.204 and 0.211 nm correspond to the (200) plane of Au and 0.235 nm is attributed to the (111) planes of Au. The selected area electron diffraction (SAED) analysis (Fig. 4c) revealed the crystalline nature of the nanoparticles in the form of three bright circular rings with lattice spacing corresponding to (111), (200) and (220) planes of the face-centered cubic lattice of gold nanoparticles.

a TEM image, b HRTEM image, and c SAED pattern of S-2 electrocatalyst
Figure 5 displays the XPS profile of the S-2 electrocatalyst. Through the XPS technology, more studies were conducted on the surface composition and chemical bonding of the S-2 composite. The XPS spectra of the Au-Co nanoparticles in the Au 4f and Co 2p regions of the S-2 catalysts are displayed in Fig. 5a and b respectively. As seen in Fig. 5a, the Au 4f7/2 peaks of the Au-Co nanoparticles in the S-2 catalysts locate at close to 84.4 eV which can be attributed to the Au 4f7/2 of the metallic gold [23]. As seen in Fig. 5b, the binding energies (BEs) of Co2+ 2p3/2 and Co2+ 2p1/2 can be attributed to the peaks at 782.1 and 798 eV respectively in the Co 2p spectrum [24]. There are two apparent shake-up satellite peaks at 787.2 and 803.7 eV. The XPS spectra of the S-2 electrocatalyst in the Au 4f and Co 2p regions are fitted using the Gaussian sum function to investigate better the surface chemical states of the Au-Co nanoparticles in the catalysts. It is observed that the S-2 electrocatalyst comprises two peaks in the Au 4f7/2 peak of the Au-Co nanoparticles, which are situated at approximately 85.1 and 84.4 eV, respectively. All of them are related to the metallic gold’s Au 4f7/2. The multielectron excitation causes two satellite peaks in the XPS spectrum of the Au-Co nanoparticles in the S-2 electrocatalyst, as seen in Fig. 5b.

XPS spectra fitted by Gaussian sum function of the AuCo nanoparticles in the S-2 catalyst in the Au 4f (a) and Co 2p (b) regions
3.2 Electrochemical performance of Aux-Co100 − x/MWCNT
Initially, CV was carried out using 0.5 M NaOH solution on our synthesized samples. Figure 6a, b shows the CV curve at a 100 mV s− 1 scan rate in 0.5 M NaOH solution at room temperature. The redox peaks were clearly visible for all electrocatalysts. The curves show distinct peaks at 0.25 V, 0.25 V, 0.22 and 0.23 V in the anodic scan for S-1, S-2, S-3 and S-4 electrodes. An identical anodic peak was observed for Co/MWCNT in the same solution (inset of Fig. 6a). These anodic peaks are accredited to the Co surface oxidation process in the sequence CoO → Co3O4 → Co2O3 [25]. At about − 0.85 V, the S-3 and S-4 modified electrode surfaces displayed additional little anodic humps. These anodic characteristics are attributed to electrochemical gold oxide layer development on electrode surfaces and \(O{H}^{-}\) adsorption [26]. The cathodic peaks on the S-2 modified electrode were located at 0.046 V and − 0.47 V. At 0.043 V and − 0.47 V, similar cathodic characteristics were seen on the surface of the S-4 electrode. Two cathodic peaks with center values of 0.05 V and − 0.42 V could be seen on the S-3 modified electrode. The S-1 catalyst showed one cathodic peak centered at 0.07 V. Reduction of hydroxides and oxides (produced in anodic scan) into elemental Au and Co is responsible for these cathodic peaks [26, 27]. According to the results, the Aux-Co100 − x/MWCNT modified electrode exhibits greater electrocatalytic activity than Au/MWCNT. As a result, the conduction of electrons in the MWCNT-supported bimetallic system is increased due to the presence of Co. Duan’s group gives a similar explanation in their reported Cu@Ag core-shell electrocatalyst towards borohydride oxidation. In their work, the presence of Cu increases the electron conduction activity of the bimetallic system [28]. Additionally, a comparison of the CVs for the Au/MWCNT and Aux-Co100 − x/MWCNT electrodes reveals a further negative sweep of potentials in the reduction peaks for the bifunctional material, indicating a stronger Oad interaction with Au surface sites in the presence of Co, which may help break the oxygen-oxygen bond [29]. This investigation confirms the electrocatalytic activities of our prepared electrocatalysts in an alkaline medium.

a, b CV of all synthesized electrocatalysts at a scan rate of 100 mV s− 1 at 303 K in a 0.5 M NaOH solution (Inset shows the CV curve of Co/MWCNT in identical condition), and c, d CV of all synthesized electrocatalysts at a scan rate of 100 mV s− 1 at 303 K in a solution of 0.5 M NaOH and 0.01 M NaBH4.
At a scan rate of 100 mV s− 1, throughout a potential range of − 1.2 to 0.8 V vs. Ag/AgCl, the borohydride oxidation reaction (BOR) kinetics of Aux-Co100 − x/MWCNT and Au/MWCNT catalysts were studied. The CV curves of all the as-prepared electrocatalysts in the presence of NaBH4 (0.01 M) in the same alkaline solution are shown in Fig. 6c, d. The cyclic voltammograms of borohydride oxidation on the Aux-Co100 − x/MWCNT electrode and borohydride oxidation on the Au/MWCNT electrode resembled each other, demonstrating that the electrooxidation mechanism of B\({H}_{4}^{-}\) on the surfaces of these electrocatalysts was the same. Two prominent oxidation peaks were found in the CV profile which matched those of earlier investigations using Au electrodes in alkaline liquids [30]. The direct oxidation of BH4− ions produced the first oxidation peak (A1) which ranged from − 0.5 V to 0 V [31]. The second anodic peak, designated A2, was created at 0.29 V as a result of the oxidation of the reaction intermediate on the partially oxidized Au surface [9]. Peak A3 on the CV curve of electrocatalyst electrodes which appears in the presence of Co, can be attributed to the catalytic oxidation of H2 caused by the hydrolysis of BH4− ion (Eqs. (3) and (4)) [32]. A sharp peak at roughly 0.35 V was visible on the reverse scan. The oxidation of absorbed species, such as BH3OH that are generated as intermediates during the oxidation of BH4− ions in the forward reaction can be credited with the formation of this peak (Eq. (5)) [31].
Then the direct oxidation of B\({H}_{4}^{-}\) obtained the A1 peak current density. S-2 displayed the highest forward peak current density (specific activity) of 24.146 mA.cm− 2 among all the electrocatalysts at a potential of − 383 mV (vs. Ag/AgCl). Table 2 provides information on forward peak current values in terms of specific activity and mass activity and corresponding potentials at 100 mV s− 1 scan rates.
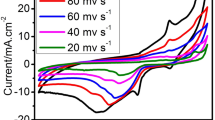
We performed CV at different scan rates in the mentioned electrolyte solution for all prepared catalysts to understand the impact of scan rate on borohydride electrooxidation performance and the results are shown in Fig. 7. The electrochemical performance is improved with the application of greater scan rates. With increased scan rates, the cathodic peak potential moves in a more cathodic direction whereas the anodic peak potential moves in a more anodic direction. The highest forward peak potentials displayed a shift in a more anodic direction with the rise in current as the cyclic voltammetry scan rates increased. The borohydride oxidation peak current density (jp) increases linearly with the square root of scan rate (inset of Fig. 7). The positive shift of the forward peak potentials and increase of jp with scan rates is a characteristic of irreversible systems [33].

a–d CV of all four electrocatalysts at different scan rates (100 mV s− 1, 80 mV s− 1, 60 mV s− 1, 40 mV s− 1 and 20 mV s− 1) in 0.5 M NaOH + 0.01 M NaBH4 solution. (Inset Shows square root of scan rates vs. current density)
To effectively execute the reaction process, it is essential to investigate the kinetics of the electrode and interfacial reactions in electrochemical systems. EIS is a powerful tool for exploring charge transfer kinetics in electrochemical reactions. Inspired by this, we conducted EIS experiments for S-1, S-2, S-3 and S-4 modified electrodes in 0.5 M NaOH + 0.01 M NaBH4 electrolyte solution at a potential of − 0.6 V (close to the anodic peak potential of borohydride oxidation) between 105 and 0.1 Hz. Figure 8 depicts the achieved Nyquist plots from the recorded EIS data. The graphs, primarily in the high-frequency region, displayed an arc-like structure. Concerning the electrochemical activity of catalysts, the diameter of the impedance arc is related to the resistance for charge transfer (Rct) in the electrolyte-electrode interface [34]. The smaller diameter of the impedance arc suggests the lower value of Rct which is essential for an electrocatalyst’s superior electron transfer kinetics and electrical conductivity. Cui et al. [35] reported the interpretations of the impedance map’s linear and semicircular portions. In the EIS profile, the high-frequency section shows a semicircular impedance arc. Its diameter is equivalent to the electron transfer resistance at the interface between electrode and electrolyte solution during borohydride oxidation. In the low-frequency range, diffusion control of Warburg impedance is indicated. In Fig. 8, no linear impedance was found at low frequencies. It was clear from this finding that it was a key factor in charge transfer at the electrode/electrolyte interface. The semicircular impedance arc validates the charge-transfer-limited process [7]. As shown in Fig. 8, the impedance spectrum of all four electrodes was fitted using resistors (Rs, Rct) and a constant phase component (CPE). Where Rs represents solution resistance, Rct represents charge transfer resistance and CPE represents the constant phase element of double-layer capacitance [7, 36]. The calculated values of Rs and Rct of all electrocatalysts are listed in Table 3. The smallest Rct value of the S-2 electrocatalyst suggests the lowest resistance for electron transfer and faster borohydride oxidation rate.

EIS spectra of all four electrocatalysts for borohydride electrooxidation at potential − 0.6 V in 0.5 M NaOH + 0.01 M NaBH4 solution. (Inset shows corresponding equivalent circuit)
The CV was examined for all electrocatalysts in a 0.5 M NaOH + 0.01 M NaBH4 solution over the temperature range of 308 to 328 K (Fig. 9). It has been observed that current density increases with an increase in temperature which indicates that the kinetics of electrochemical reactions accelerate with temperature. However, CV at 333 K temperature showed a slight decrease that the experiment’s constant fuel use might have brought. Again, at a high temperature, the rate of NaBH4 hydrolysis increases producing more H2 gas [37, 38]. The H2 gas disperses away from the electrode surface, which could affect the electrolyte’s stability and may burden the electron transfer. The Arrhenius equation (Eq. (6)) was used to determine the apparent activation energy (Eapp) of all prepared catalysts in 0.5 M NaOH + 0.01 M NaBH4 solutions.

a–d CV curve of all four electrocatalysts at different temperatures (308 K, 313 K, 318 K, 323 K, and 328 K) in 0.5 M NaOH + 0.01 M NaBH4 solution. (Inset shows the corresponding logj versus T− 1 (K− 1))
where j, T and R are the current density (mA.cm− 2), temperature (Kelvin) and molar gas constant (8.314 J/mol.K) respectively. The inset of all CV curves represents the Arrhenius plot (log j vs. 1/T). The Eapp of all electrocatalysts in a 0.5 M NaOH + 0.01 M NaBH4 electrolyte solution was evaluated using this slope value of Arrhenius plot and found to be 21.97 kJmol− 1, 8.22 kJmol− 1, 8.47 kJmol− 1 and 8.32 kJmol− 1. The values of Eapp are lower than the reported values of different electrocatalysts such as Ag-Co/C and Co-Bi/CNT (31.86 and 36.774 kJmol− 1) [5, 35]. From our investigation, it is clear that AuxCo100 − x/MWCNT electrocatalysts have lower Eapp value than Au/MWCNT indicating the improved electrocatalytic performance of the Au-Co bimetallic system.
The chronoamperometry measurement offers further details regarding the electroactivity and stability of the electrocatalysts for B\({H}_{4}^{-}\) oxidation. The chronoamperometric response was examined for the S-2-modified electrode at 0.2 V in 0.5 M NaOH + 0.01 M NaBH4 solution within a temperature range of 303 to 328 K (Fig. 10a). The Cottrell equation (Eq. (7)) can be used to analyze the CA data to determine the exchange electron number (n), which is a crucial parameter in an electrochemical reaction [39].

a CA curve of S-2 electrocatalyst in 0.5 M NaOH + 0.01 MNaBH4 solution at different temperatures. b A plot of current density versus t− 1/2 from CA data of S-2 electrocatalyst in 0.5 M NaOH + 0.01 M NaBH4 solution
where F, C and D are the Faraday’s constant (96,485 C mol− 1), B\({H}_{4}^{-}\) concentration, and diffusion coefficient. The values of D for various NaOH concentrations and temperatures, taking into account that D is independent of B\({H}_{4}^{-}\) concentration, was reported by Wang et al. [40]. For the S-2-tailored electrode in 0.5 M NaOH + 0.01 M NaBH4 solution at room temperature, the slope value of the j versus t− 1/2 plot (Fig. 10b) was determined to be 11.0261. The n value for the S-2 electrocatalyst in 0.5 M NaOH + 0.01 M NaBH4 solution at 303 K was estimated as 4.7 using this slope value and the Cottrell equation (Eq. (7)). The other reported values of n for BOR at Co1-Au1/C, Co2-Au1/C, Co4-Au1/C, and Co6-Au1/C electrodes are 0.6, 0.8, 4.1, and 3 respectively [7]. When n is less than 8, B\({H}_{4}^{-}\) has only undergone modest anodic oxidation, with the loss of available electrons mainly caused by B\({H}_{4}^{-}\) hydrolysis.
CV was examined at the S-2 electrode at a variety of NaBH4 concentrations (0.01 M to 0.08 M) to investigate the impact of NaBH4 concentration on the catalytic performance of the S-2 electrocatalyst for borohydride oxidation. Figure 11 illustrates the CV profile (100 mV s− 1 scan rate) of the S-2 electrocatalyst at various NaBH4 concentrations. The CV profile demonstrated that increasing the concentration of NaBH4 led to a substantial increase in forwarding current densities, demonstrating the electrocatalytic strength of the S-2 electrocatalyst for borohydride electrooxidation. Table 4 summarizes the forward current densities and corresponding peak positions of the S-2 electrocatalyst. We noticed a significant increase in current up to an increment of 0.06 M of NaBH4 concentration. However, it was noted that the current started to decrease when NaBH4 concentration became higher (0.07 M). This is due to a relatively high concentration of NaBH4 that continues to be consumed on the electrode surface beyond 0.06 M. Because of this, the examined electrode’s electrochemical kinetics may be disturbed. Additionally, with greater NaBH4 concentrations, we observed a positive shift in peak potentials.

CV curve of S-2 electrocatalyst in 0.5 M NaOH + x M NaBH4 (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) at 100 mV s− 1 scan rate
To check the stability of the S-2 electrocatalyst, CV up to 300 cycles was performed at 100 mV s− 1 scan rate at room temperature in 0.5 M NaOH + 0.01 M NaBH4 solution. In Fig. 12, the S-2 electrocatalyst shows the stability profile. From the 80th cycle, we observed almost uniform current densities up to 260 cycles indicating excellent stability of our synthesized electrocatalyst. After that, we observed a slight current decay due to the continuous fuel consumption on the electrode surface.

Stability of S-2 electrocatalyst in 300 scans
4 Conclusion
In the present research work, a reduction technique in an aqueous solution was used to synthesize multiwalled carbon nanotubes supported by gold-cobalt (Au-Co/MWCNT) nanoparticles with four distinct Au/Co ratios. The synthesized electrocatalysts exhibited effective electrocatalytic activity for borohydride oxidation. The findings of the experiments exhibited that the Au-Co/MWCNT catalyst had better NaBH4 oxidation kinetic parameters and transfer resistance than the Au/C catalyst, suggesting that the addition of cobalt to Au/MWCNT can effectively enhance the catalytic activity of bimetallic catalysts. In terms of current density and charge transfer resistance, the S-2 electrocatalysts performed electrocatalytically better than S-1, S-3 and S-4 as an anode in DBFC. In addition, in S-2 as-prepared electrocatalyst, the average size of particles was 4.1 nm. In this investigation, the maximum current density at room temperature, charge transfer resistance, number of exchange electrons and apparent activation energy of the S-2-tailored electrode were calculated as 24.146 mA.cm− 2, 134.9 Ω, 4.7 and 8.22 kJmol− 1 respectively. Thus, this Au-Co/MWCNT bimetallic electrocatalyst can be potentially used as a new type of anode electrocatalyst for borohydride electrooxidation.
Data availability
Data will be made available on reasonable request.
References
de Leon CP, Walsh FC, Rose A, Lakeman JB, Browning DJ, Reeve RW (2007) A direct borohydride—acid peroxide fuel cell. J Power Sour 164:441–448. https://doi.org/10.1016/j.jpowsour.2006.10.069
Cheng H, Scott K (2006) Influence of operation conditions on direct borohydride fuel cell performance. J Power Sour 160:407–412. https://doi.org/10.1016/j.jpowsour.2006.01.097
Atwan MH, Macdonald CL, Northwood DO, Gyenge EL (2006) Colloidal au and Au-alloy catalysts for direct borohydride fuel cells: electrocatalysis and fuel cell performance. J Power Sour 158(1):36–44. https://doi.org/10.1016/j.jpowsour.2005.09.054
Li ZP, Liu BH, Arai K, Asaba K, Suda S (2004) Evaluation of alkaline borohydride solutions as the fuel for fuel cell. J Power Sour 158:36–44. https://doi.org/10.1016/j.jpowsour.2003.08.017
Dey S, Pramanik S, Chakraborty P, Rana DK, Basu S (2022) An easy synthesis of carbon-supported silver–cobalt bimetallic nanoparticles to study the electrocatalytic performance in alkaline borohydride fuel cell. J Appl Electrochem 52:247–258. https://doi.org/10.1007/s10800-021-01641-2
Indig ME, Snyder RN (1962) Sodium borohydride, an interesting anodic fuel(1). J Electrochem Soc 109(11):1104. https://doi.org/10.1149/1.2425247
Duan D, Feng J, You X, Zhou X, Wang Y, Chen L, Liu S (2021) Evaluation of Co–Au bimetallic nanoparticles as anode electrocatalyst for direct borohydride-hydrogen peroxide fuel cell. Ionics 27:3521–3532. https://doi.org/10.1007/s11581-021-04115-9
Yi L, Liu L, Liu X, Wang X, Yi W, He P, Wang X (2012) Carbon-supported Pt–Co nanoparticles as anode catalyst for direct borohydride-hydrogen peroxide fuel cell: electrocatalysis and fuel cell performance. Int J Hydrog Energy 37:12650–12658. https://doi.org/10.1016/j.ijhydene.2012.06.065
Gyenge E (2004) Electrooxidation of borohydride on platinum and gold electrodes: implications for direct borohydride fuel cells. Electrochim Acta 49:965–978. https://doi.org/10.1016/j.electacta.2003.10.008
Xu CW, Wang H, Shen PK, Jiang SP (2007) Highly ordered pd nanowire arrays as effective electrocatalysts for ethanol oxidation in direct alcohol fuel cells. Adv Mater 19(23):4091–4304. https://doi.org/10.1002/adma.200602911
Zhang Y, Janyasupab M, Liu CW, Li X, Xu J, Liu CC (2012) Three dimensional PtRh alloy porous nanostructures: tuning the atomic composition and controlling the morphology for the application of direct methanol fuel cells. Adv Funct Mater 22(17):3570–3575. https://doi.org/10.1002/adfm.201200678
Evans GE, Kordesch KV (1967) Hydrazine-air fuel cells: hydrazine-air fuel cells emerge from the laboratory. Science 158(3805):1148–1152. https://doi.org/10.1126/science.158.3805.1148
Han SB, Song YJ, Lee YW, Ko AR, Oh JK, Park KW (2011) High-performance hydrogen fuel cell using nitrate reduction reaction on a non-precious catalyst. Chem Commun 47(12):3496–3498. https://doi.org/10.1039/C0CC05534D
Yu SS, Lee TH, Oh TH (2022) Ag–Ni nanoparticles supported on multiwalled carbon nanotubes as a cathode electrocatalyst for direct borohydride–hydrogen peroxide fuel cells. Fuel 315:123151. https://doi.org/10.1016/j.fuel.2022.123151
Santos DMF, Sequeira CAC (2010) Cyclic voltammetry investigation of borohydride oxidation at a gold electrode. Electrochim Acta 55:6775–6781. https://doi.org/10.1016/j.electacta.2010.05.091
Nagle LC, Rohan JF (2011) Nanoporous gold anode catalyst for direct borohydride fuel cell. Int J Hydrog Energy 36(16):10319–10326. https://doi.org/10.1016/j.ijhydene.2010.09.077
Martins M, Šljukić B, Metin Ö, Sevim M, Sequeira CA, Şener T, Santos DM (2017) Bimetallic PdM (M = fe, ag, au) alloy nanoparticles assembled on reduced graphene oxide as catalysts for direct borohydride fuel cells. J Alloys Compd 718:204–214. https://doi.org/10.1016/j.jallcom.2017.05.058
Oh TH (2021) Gold-based bimetallic electrocatalysts supported on multiwalled carbon nanotubes for direct borohydride–hydrogen peroxide fuel cell. Renew Energy 163:930–938. https://doi.org/10.1016/j.renene.2020.09.028
He P, Wang X, Liu Y, Liu X, Yi L (2012) Comparison of electrocatalytic activity of carbon-supported Au–M (M = fe, Co, Ni, Cu and Zn) bimetallic nanoparticles for direct borohydride fuel cells. Int J Hydrog Energy 37(16):11984–11993. https://doi.org/10.1016/j.ijhydene.2012.05.054
Oh TH, Jang B, Kwon S (2014) Electrocatalysts supported on multiwalled carbon nanotubes for direct borohydride–hydrogen peroxide fuel cell. Int J Hydrog Energy 39(13):6977–6986. https://doi.org/10.1016/j.ijhydene.2014.02.117
Yi L, Wei W, Zhao C, Tian L, Liu J, Wang X (2015) Enhanced activity of Au–Fe/C anodic electrocatalyst for direct borohydride-hydrogen peroxide fuel cell. J Power Sour 285:325–333. https://doi.org/10.1016/j.jpowsour.2015.03.118
He P, Wang X, Fu P, Wang H, Yi L (2011) The studies of performance of the au electrode modified by Zn as the anode electrocatalyst of direct borohydride fuel cell. Int J Hydrog Energy 36(15):8857–8863. https://doi.org/10.1016/j.ijhydene.2011.04.128
Yan S, Gao L, Zhang S, Gao L, Zhang W, Li Y (2013) Investigation of AuNi/C anode catalyst for direct methanol fuel cells. Int J Hydrog Energy 38(29):12838–12846. https://doi.org/10.1016/j.ijhydene.2013.07.102
Yin D, Tang J, Bai R, Yin S, Jiang M, Kan Z, Li H, Wang F, Li C (2021) Cobalt phosphide (Co2P) with notable electrocatalytic activity designed for sensitive and selective enzymeless bioanalysis of hydrogen peroxide. Nanoscale Res Lett 16(1):1–10. https://doi.org/10.1186/s11671-020-03469-9
Pourbaix M (1974) Atlas of electrochemical equilibria in aqueous solution. NACE
Pei F, Wang Y, Wang X, He P, Chen Q, Wang X, Wang H, Yi L, Guo J (2010) Performance of supported Au–Co alloy as the anode catalyst of direct borohydride-hydrogen peroxide fuel cell. Int J Hydrog Energy 35(15):8136–8142. https://doi.org/10.1016/j.ijhydene.2010.01.016
Duan D, Liang J, Liu H, You X, Wei H, Wei G, Liu S (2015) The effective carbon supported core–shell structure of Ni@ au catalysts for electro-oxidation of borohydride. Int J Hydrog Energy 40(1):488–500. https://doi.org/10.1016/j.ijhydene.2014.10.101
Duan D, Liu H, You X, Wei H, Liu S (2015) Anodic behavior of carbon supported Cu@ Ag core–shell nanocatalysts in direct borohydride fuel cells. J Power Sour 293:292–300. https://doi.org/10.1016/j.jpowsour.2015.05.086
Wang Y, Lu X, Liu Y, Deng Y (2013) Silver supported on Co3O4 modified carbon as electrocatalyst for oxygen reduction reaction in alkaline media. Electrochem Commun 31:108–111. https://doi.org/10.1016/j.elecom.2013.03.017
Yang F, Cheng K, Wang G, Cao D (2015) Preparation of au nanosheets supported on ni foam and its electrocatalytic performance towards NaBH4 oxidation. Electrochim Acta 159:111–115. https://doi.org/10.1016/j.electacta.2015.01.171
Tamašauskaitė-Tamašiūnaitė L, Balčiūnaitė A, Šimkūnaitė D, Selskis A (2012) Self-ordered titania nanotubes and flat surfaces as a support for the deposition of nanostructured Au–Ni catalyst: enhanced electrocatalytic oxidation of borohydride. J Power Sour 202:85–91. https://doi.org/10.1016/j.jpowsour.2011.11.030
Chen D, Liu S, Li J, Zhao N, Shi C, Du X, Sheng J (2009) Nanometre Ni and core/shell Ni/Au nanoparticles with controllable dimensions synthesized in reverse microemulsion. J Alloys Compd 475(1–2):494–500. https://doi.org/10.1016/j.jallcom.2008.07.115
Dey S, Chakraborty P, Rana DK, Pramanik S, Basu S (2021) Surfactant-free synthesis of carbon-supported silver (Ag/C) nanobars as an efficient electrocatalyst for alcohol tolerance and oxidation of sodium borohydride in alkaline medium. SN Appl Sci 3(6):1–12. https://doi.org/10.1007/s42452-021-04601-9
Hansu TA, Caglar A, Sahin O, Kivrak H (2020) Hydrolysis and electrooxidation of sodium borohydride on novel CNT supported CoBi fuel cell catalyst. Mater Chem Phys 239:122031. https://doi.org/10.1016/j.matchemphys.2019.122031
Cui N, Luo JL (1998) Effects of oxide additions on electrochemical hydriding and dehydriding behavior of Mg2Ni-type hydrogen storage alloy electrode in 6 M KOH solution. Electrochim Acta 44:711–720. https://doi.org/10.1016/S0013-4686(98)00235-7
Abdel-Rehim SS, Khaled KF, Abd-Elshafi NS (2006) Electrochemical frequency modulation as a new technique for monitoring corrosion inhibition of iron in acid media by new thiourea derivative. Electrochim Acta 51(16):3269–3277. https://doi.org/10.1016/j.electacta.2005.09.018
Liu BH, Li ZP (2009) A review: hydrogen generation from borohydride hydrolysis reaction. J Power Sour 187(2):527–534. https://doi.org/10.1016/j.jpowsour.2008.11.032
Zhang D, Ye K, Cheng K, Cao D, Yin J, Xu Y, Wang G (2014) High electrocatalytic activity of cobalt–multiwalled carbon nanotubes–cosmetic cotton nanostructures for sodium borohydride electrooxidation. Int J Hydrog Energy 39(18):9651–9657. https://doi.org/10.1016/j.ijhydene.2014.04.113
Birry L, Lasia A (2004) Studies of the hydrogen evolution reaction on raney nickel—molybdenum electrodes. J Appl Electrochem 34(7):735–749. https://doi.org/10.1023/B:JACH.0000031161.26544.6a
Wang K, Lu J, Zhuang L (2005) Direct determination of diffusion coefficient for borohydride anions in alkaline solutions using chronoamperometry with spherical au electrodes. J Electroanal Chem 585(2):191–196. https://doi.org/10.1016/j.jelechem.2005.08.009
Acknowledgements
The authors acknowledge the centre of excellence (COE), NIT Durgapur, for the FESEM facility.
Funding
The authors acknowledge the DST&BT project (Project no. 326(Sanc.)/ST/P/S&T 16G-21/2018), Govt. of West Bengal, for financial support during this work.
Author information
Authors and Affiliations
Contributions
CKR: Investigation, Formal analysis, Writing—original draft, Validation. SD: Resources, Formal analysis, Validation. MH: Writing—original draft, Validation. RK: Resources, Validation. SB: Resources, Validation AKM: Supervision, Conceptualization, Methodology, Visualization, Validation, Project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This declaration is “not applicable”.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Raul, C.K., Dey, S., Halder, M. et al. Synthesis of AuxCo100 − x/MWCNT nanoparticles as an efficient anode electrocatalyst for borohydride oxidation in alkaline medium. J Appl Electrochem 53, 977–990 (2023). https://doi.org/10.1007/s10800-022-01824-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-022-01824-5