
Abstract
Anodic electrodeposition of lead dioxide at the bare and Nafion® covered gold electrode was studied by cyclic voltammetry and chronoamperometry. The results showed that Nafion® layer has a favourable effect on the efficiency of the deposition process which was caused by both thermodynamic and kinetic reasons. Electrodeposition process at Nafion® covered gold electrode goes via Pb(III) intermediate species which are stabilized within Nafion® membrane. The final PbO2 deposit crystallizes in tetragonal β-PbO2 form.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Anodic deposition of lead (IV) oxide at solid support was one of the most thoroughly investigated electrochemical processes. Huge research efforts have been made in order to elucidate the mechanism of electrodeposition, its kinetics and relationship between deposition conditions and structure of the resulting PbO2 layers. These researches were driven by at least two strong motivations. First, studies of electrochemical deposition of PbO2 gained insight into the nucleation and growth process and its kinetics which enabled the development of the general theoretical nucleation and growth theory which could be applied not only to understand subtle details of PbO2 electrodeposition but also for the electrocrystallization processes of other oxide layers as well as cathodically formed metal deposits. Indeed, experimental results and their analysis in the works of Fleishmann [1–6] and later by Abyaneh [7–10], and others [11–16] paved the way toward the development of the 2D/3D instantaneous and progressive nucleation processes.
Another incentive for studying PbO2 electrodeposition lies in its interesting electrochemical and catalytic properties which opened the ways for the broad applications of this material especially as cathode material in lead acid batteries [17–19] and as catalytic material in various electrochemical and chemical processes [20–23].
Recently, PbO2 was considered as an active electrode material in hybrid supercapacitors [24, 25], devices assembled as a combination of one double layer charging (DLC) electrode and another pseudocapacitive electrode. Its high standard reduction potential, very facile electrochemical kinetics and high conductivity make PbO2 very attractive for the development of high energy/high power supercapacitor devices.
In order to increase charging/discharging rate of PbO2 electrodes which is required in high-power applications and at the same time keeping its high energy content, it is of crucial importance to prepare highly porous nano-structured PbO2 layer which will enable facile ionic charge transport, especially sulphate ions and protons, into and through the PbO2 layer.
In this work, our idea was to study anodic template deposition of PbO2 at Nafion® covered solid support, in our case gold electrode. Interesting physico-chemical properties of Nafion membrane give rise to many applications of this material including PEM fuel cells [26, 27], energy and power tuning in pseudocapacitive materials [28, 29], PEM technology electrolysis cells [30] and as matrix for entrapment of biomolecules [31]. The PbO2 electrodeposition was expected to be governed by the slow diffusion of Pb(II) ions through Nafion® micro- and nano-channels toward the gold support. Anodic oxidation of Pb(II) should have yielded nano-clustered PbO2 layer with the structure which would depend on the type of Nafion® used, its porosity and content of hydrophilic groups.
The main goal of this work was not completely achieved but the results we obtained were sufficiently interesting to be demonstrated in this paper. Up to our knowledge, no results describing PbO2 deposition at gold electrode covered by Nafion® layer exist in the literature. There were, however, a couple of papers dealing with PbO2 electrodeposition from the solutions of Pb(II) ion in the presence of dissolved Nafion® [32, 33]. The results of these investigations were very interesting and showed that the presence of Nafion® did not hinder the electrodeposition process but rather facilitated it.
Experimental
Materials
All chemicals used in this work were of analytical grade and they were purchased: Pb(NO3)2 from Alfa Aesar, HNO3 from Sigma Aldrich and Nafion® 117 Solution (5 %—in mixture of lower aliphatic alcohols and water) from Fluka. All solutions were prepared from double-distilled water.
Preparation of gold and Nafion® covered gold electrode
Electrodeposition of PbO2 was performed at two types of working electrodes: the bare gold electrode and the gold electrode covered with Nafion® layer. The surface area of gold electrode was 0.07 cm2. Before electrodeposition, gold electrodes were polished with Al2O3 slurry, cleaned with ethanol in ultrasonic bath for 10 min and washed with double distilled water. The Nafion® covered gold electrodes were prepared by casting of Nafion® solution. In order to get several different thickness of Nafion® layers (10, 60, 120, 300 μm), 5 % Nafion® solution was diluted with double-distilled water. The Nafion® covered gold electrodes were dried for 1 h at room temperature and open air.
Electrochemical measurements
Electrochemical measurements were performed in three-compartment cell by means of an Autolab Potentiostat/Galvanostat PGSTAT100. Three-electrode system contained working electrode: the bare gold or the Nafion® covered gold electrode, Pt-foil counter electrode and Ag/AgCl (3 M KCl) reference electrode. Electrodepositon of PbO2 was performed from solution of 0.01 M Pb(NO3)2 in 1 M HNO3, using method of cyclic voltammetry in the potential range from 0 to 1.8 V at scan rate of 50 mV/s. The same solution was used for electrodepostion of PbO2 at gold rotating disk electrode (RDE) with the method of chronoamperometry at potential of 1.55 V during 200 s and the rotation rate of 1000 rpm. The RDE gold electrode was also either bare or covered with 120 μm of thick Nafion® layer. Mechanism of PbO2 electrodeposition was monitored using current-time transients at several different potentials: 1.6, 1.65, 1.7, 1.75 and 1.8 V during period of 20 s. All potentials in this paper are referred to Ag/AgCl reference electrode.
Characterization
The morphology and the size of electrodeposited PbO2 and PbO2 /Nafion® layer were examined by scanning electron microscope (SEM) Vega 3 SEM TESCAN at the efficient voltage of 20 kV.
Structural features of electrodeposited PbO2 and PbO2 /Nafion® layer were studied by the X-ray powder diffraction (XRPD) at room temperature using a Philips 1830 counter diffractometer with Cu Kα radiation. Samples were recorded in 2ϑ range 10–70°, with the measuring step of 0.02° and fixed counting time 1 s/step.
Results and discussion
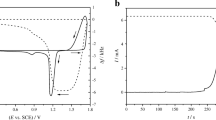
Cyclic voltammograms of PbO2 electrodeposition from 0.01 M Pb(NO3)2 at a bare gold electrode and at a gold electrode covered with Nafion® are shown in Fig. 1. The cyclic voltammograms at bare gold electrode exhibit familiar shape which consists of several well-defined current peaks, both in anodic and cathodic potential excursion. Pb(II) electrooxidation takes place at the potentials of the current peak of about 1.65 V followed by the current rise which is the result of oxygen evolution reaction. In the cathodic branch of the voltammograms, two current peaks appear corresponding to the dissolution of lead and gold oxides at about 1.1 and 0.75 V, respectively.

Cyclic voltammogram of PbO2 electrodeposition from 0.01 M Pb (NO3)2/1 M HNO3 solution at the bare gold electrode (black line) and the gold electrode covered with Nafion® (red dashes). Gold electrode in background electrolyte (blue trace). Scan rate of 50 mV s−1
Mechanism of Pb(II) electrooxidation and PbO2 electrodeposition involves the nucleation and growth process as described in the numerous papers dealing with the subject [1–16]. From these investigations, it is now well established that the PbO2 electrodeposition is a complex process which involves as a first step a mediated oxidation of Pb(II) ion via adsorbed hydroxyl radicals which are formed at the gold electrode:
The hydroxyl radicals are involved in Pb(II) oxidation resulting in the soluble oxygen containing intermediate lead species. However, despite the huge amount of data, it is still the matter of debate what is the nature of these intermediates. One group of authors suggests that Pb(III) species are formed according to the following mechanism [34]:
while another group of authors speculate the formation of soluble Pb(IV) as intermediate [1, 2, 35]:
In both cases, the formation of nuclei seems to be the critical factor in controlling the overall electrodeposition process. When electrodeposition takes place at a bare gold electrode (black curve, Fig. 1), substantial nucleation overpotential was observed as a difference between the potential of the onset of anodic current in the forward scan and the equilibrium potential, i.e. potential where the current trace intersects potential axis in the reverse scan. Depending on the experimental conditions, nucleation overpotential varies between 150 and 200 mV.
Counterintuitive feature of the cyclic voltammogram obtained at the gold electrode covered with Nafion® is unexpectedly high registered currents at the potentials of PbO2 electrodeposition which indicates that Nafion® does not inhibit electrode reaction by blocking the active sites at the surface but rather facilitates it. Another interesting feature of the cyclic voltammograms at the Nafion® covered gold electrode is the diminishing of the nucleation overpotential since the onset of the oxidation current rise coincides with the equilibrium potential of PbO2. Obviously, the formation of nuclei at Nafion® covered electrode is more favourable than at bare gold electrode which might be caused by both thermodynamic and kinetic factors. Indeed, in the inset of Fig. 1, it could be discerned that the current rise starts even before the PbO2 equilibrium potential resembling the phenomena usually observed in underpotential depositions of species at foreign substrates. The occurrence of underpotential deposition effect in this case reveals favourable energetics of the PbO2 nuclei formation within Nafion® membrane which is most probably due to the strong interactions of soluble lead intermediate species with the polar groups on the polymer chains.
In his studies of the effect of dissolved Nafion® [32] and the effect of other fluorine containing compounds [33] on PbO2 electrodeposition, Velichenko identified at least two types of interactions which might exist between Nafion® and lead compounds and affect electrodeposition kinetics and the properties of the resulting layers. Apart from the bulk effects caused by the interaction of positive lead ions with Nafion® polyanionin bulk electrolyte, he found that the adsorption of polyelectrolyte on PbO2 occurs not only due to the electrostatic attraction of polyanion to the positively charged surface of PbO2 but also the existence of some specific interactions. As the result, the increase of electron transfer rates with the increase of Nafion® concentration was observed up to the critical Nafion® concentrations where blocking of the electrode by Nafion® became dominant factor in the control of the reaction.
In our work, the effect of dissolved Nafion® on the PbO2 electrodeposition is not so pronounced as described in Velichenko’s papers (results not shown). The only noticeable difference between cyclic voltammograms obtained at bare gold electrode with and without dissolved Nafion® is a decreased height of the cathodic current peak corresponding to PbO2 reduction for about 15 % in the case when Nafion® is present in the solution. This feature confirms the existence of the bulk interactions between soluble lead intermediate species with Nafion® polyelectrolyte. The interactions are most likely of electrostatic origin.
The cyclic voltammetric results obtained with Nafion® covered gold electrode offered several new and important insights in the overall process of PbO2 electrodeposition. It was demonstrated that Nafion® layer does not hinder neither the diffusion of Pb2+ ion toward the electrode nor the electron transfer at gold/Nafion® interface. Oxygen reactive species formed at the electrode as a result of the reaction (1) are more stable and longer living species within the Nafion® environment than at the bare gold electrode. Consequently, PbO2 electrodeposition takes place throughout the whole volume of Nafion® membrane and therefore the current efficiency is increased resulting in the faster PbO2 nucleation kinetics. If the oxidation of Pb2+ ion would proceed directly at the gold electrode, the diminishing of the oxidation current could be expected as is the case in the electrodeposition of NiOOH from Ni2+ solutions (Fig. 2).

Cyclic voltammogram of NiOOH electrodeposited from 0.1 M NiSO4 solution at the bare gold electrode (black line) and at the gold electrode covered with Nafion® layer (red dash). Scan rate of 5 mV s−1
Additionally, these results are in favour of the postulated deposition mechanism which goes via Pb(III) soluble species as described in Eqs. 2 and 3, while the mechanism involving soluble Pb(IV) species (Eq. 4) can be excluded due to energetically unfavourable formation of negatively charged hydroxyl ions in the cation-exchanging polymer matrix.
Most frequently used and widely accepted method for the studying mechanism and kinetics of the nucleation and growth processes of the formation of solid layers at electrodes is recording the current-time transients upon potential step. Depending on the applied potential, nature of solid support and electrolyte used, current-time transients for PbO2 formation can assume a sigmoidal shape [34–36] or a current peak can be formed. Current peaks were usually observed during 2D or 3D nucleation with diffusionally controlled growth process [37–39]. Sometimes, a growth inhibition was identified and described [40].
Figure 3 shows the current-time transients obtained from the solution of Pb2+ at bare (Fig. 3a) and Nafion® covered gold electrode (Fig. 3b) at different potentials. In both cases, familiar shapes were observed with the initial current spike followed by the well-formed current peaks and the final current decay toward steady state values. As might be expected for the transients representing nucleation and growth process, registered current peak maxima shift toward lower times and their height increase as the deposition potential increases. The current decay immediately after the peaks shows Cottrell linearity revealing diffusional control of the reaction at this stage. However, current values at longer times coincide for all potentials investigated indicating a transition from diffusional to kinetic control which is most likely the result of growth inhibition as described in [40]. The increased current at the potentials higher than 1.8 V is due to contribution of the oxygen evolution reaction. In accordance with cyclic voltammetric measurements, currents registered at the Nafion® covered gold electrode are much higher than those recorded at the bare gold electrode. The position and height of the current maximum depends on the Nafion® layer thickness (Fig. 4). However, at sufficiently long times after the maximum current reaches the level it had at the bare gold electrode indicating the similarity of both processes in the advanced stage of electrodeposition.

Current-time transients at different potentials obtained from the 0.01 M Pb(NO3)2/1 M HNO3 solution at a the bare gold and b the gold electrode covered with 10-μm-thick Nafion®

Current-time transients at potential of 1.6 V obtained from the 0.01 M Pb(NO3)2/1 M HNO3 solution at the gold electrodes covered with different thickness of Nafion® layer: 10, 60 and 300 μm
As demonstrated by the voltammetric and chronoamperometric measurements, higher efficiency of the electrodeposition process at Nafion® covered gold electrode is the result of several factors, all of them simultaneously affecting in a greater or lesser extent the course of the reaction:
-
(i)
favourable interaction between positive lead-oxygen intermediate species and Nafion® negatively charged sulfonic groups,
-
(ii)
more favourable energetics of PbO2 nuclei formation in Nafion® environment comparing to the aqueous medium,
-
(iii)
Nafion® acting as a “cage” for soluble lead species preventing them to diffuse away into the bulk of the solution before their further oxidation and deposition.
The last factor was confirmed by the measurements taken at the rotating disk gold electrode (Fig. 5). If the measurements were performed at bare gold electrode then the rotation slowed down the rate of the electrodeposition by prolonging an induction time which was the result of enhanced transport of soluble intermediates toward the bulk solution. At Nafion® covered electrode, however, the opposite effect was observed, the current even increases comparing to the current registered in unstirred solution, and remains high and constant for a long time. The height of the current plateau is not significantly affected by the rotation rate (results not shown). Once the first layer of solid PbO2 is formed further growth is initially controlled by the diffusion of Pb2+ toward the electrode. At the sufficiently long times Nafion® pores are filled up completely by PbO2, the Nafion®/PbO2 composite material is formed, and at this stage the electrodeposition in both cases is kinetically controlled. The excess oxidation charge registered in the current transients in Fig. 4 is related to the PbO2 electrodeposited inside the whole volume of the Nafion®. This is confirmed by the constant value of the ratio of the excess charge obtained from current transients at Nafion® covered gold electrode in the time span where differences in current of two transients exist and the mass of casted Nafion® membrane at the electrode.

Electrodeposition of PbO2 from 0.01 M Pb(NO3)2/1 M HNO3 on the rotating disk gold electrode (RDE), bare (black and red curves) and covered with 120-μm-thick Nafion® layer (blue and green curves), 1000 rpm, E dep = 1.55 V
Figure 6 shows the comparison of SEM micrographs of the PbO2 deposited at gold and inside the Nafion®. It is visible that the growth of PbO2 outside Nafion®, once the membrane was filled with PbO2, results in globular structures which protrude from the pores of the Nafion®. In the case of PbO2 at gold electrode, the growth is more uniform although some globular structures could also be discerned.

SEM micrographs of the PbO2 film at magnification of a ×1000 and b ×5000 and PbO2/Nafion® film of 120 μm thickness at magnification of c ×1000 and d ×5000
Figure 7 shows XRPD patterns of PbO2 deposited at the bare Au electrode (Fig. 7a) and PbO2 deposited at the Au electrode covered with 120-μm-thick Nafion® (Fig. 7b). Diffraction lines at 2ϑ = 25, 32, 36, 49, 54, 59, 61, 63 i 67° were noticed in both XRPD patterns which points out that the lead oxide crystallizes as tetragonal β-PbO2 (with refined unit-cell parameters a = 5.003 Å, c = 3.385 Å), regardless of the electrode type used for the electrodeposition. No other polymorphic forms of PbO2 have been noticed in the sample as well as any additional impurity phases. Intense diffraction line at 2ϑ = 28° corresponds to silicon holder since a very limited amount of material has been used for diffraction data collection.

X-ray diffraction patterns of a electrodeposited PbO2 with presence of 120-μm-thick Nafion® layer and b electrodeposited PbO2 film without Nafion® layer
Conclusions
Nafion® layer at gold electrode has favourable influence on PbO2 electrodeposition from both thermodynamic and kinetic standpoints. The electrodeposition follows similar nucleation and growth mechanism as in the case of PbO2 electrodeposition at the bare gold electrode. However, the presence of Nafion® considerably diminishes nucleation overpotential resulting in the higher efficiency of the complete electrode reaction. Several factors contribute to the rapid kinetics of the process including favourable interaction between positive lead-oxygen intermediate species and Nafion® negatively charged sulfonic groups as well as more favourable energetics of PbO2 nuclei formation in Nafion® environment comparing to the aqueous medium. PbO2 layer crystallizes in tetragonal β-PbO2form.
The results obtained in this work could be applied for the construction of high efficiency and high power positive electrodes in lead acid batteries and supercapacitors.
References
Fleischmann M, Mansfield JR, Thirsk HR, Wilson HG (1967) Electrochim Acta 12:967–982
Fleischmann M, Liler M (1958) Trans Faraday Soc 54:1370–1381
Fleischmann M, Thirsk HR (1955) Trans Faraday Soc 51:71–95
Bewick A, Fleischmann M, Thirsk HR (1962) Trans Faraday Soc 58:2200–2216
Armstrong RD, Fleischmann F, Thirsk HR (1966) J Electroanal Chem 11:208–223
Fleishmann M, Thirsk HR (1963) In: Delahay P (Ed) advances in electrochemistry and chemical engineering, vol 3, Chap. 3. Wiley Interscience, New York
Abyaneh MY (1982) Electrochim Acta 27:1329–1334
Abyaneh MY (1986) J Electroanal Chem 209:1–10
Abyaneh MY (1991) Electrochim Acta 36:727–732
Abyaneh MY, Saez V, Gonzalez-Garcia J, Mason TJ (2010) Electrochim Acta 55:3572–3579
Bosco E, Rangarajan SK (1982) J Electroanal Chem 134:225–241
Gunawardena GA, Hills GJ, Montenegro I, Scharifker BR (1982) J Electroanal Chem 138:225–239
Scharifker BR, Mostany J (1984) J Electroanal Chem 177:13–23
Sluyters-Rehbach M, Wijenberg JHOJ, Bosco E, Sluyters JH (1987) J Electroanal Chem 236(1987):1–20
Mirkin MV, Nilov AP (1990) J Electroanal Chem 283:35–51
Harrinson JA, Thirsk HA (1972) In: A guide to the study of the electrode kinetics, chap. 3, Academic Press, New York.
Pavlov D, Balkanov I, Halachev T, Rachev P (1989) J Electrochem Soc 136:3189–3197
Ruetschi P (1992) J Electrochem Soc 139:1347–1351
Rand DAJ (1987) J Power Sources 21:B1
Hui BS, Huber CO (1982) Anal Chim Acta 134:211–218
Yeo IH, Kim S, Jacobson RA, Johnson DC (1989) J Electrochem Soc 136:1395–1401
Polcaro AM, Palmas S, Renoldi F, Mascia M (1999) J Appl Electrochem 29:147–151
Tahar NB, Savall A (1999) J New Mater Electrochem Syst 2:19–26
Wang F, Xiao S, Hou Y, Hu C, Liu L, Wu Y (2013) RSC Adv 3:13059–13084
Simon P, Gogotsi Y (2008) Nat Mater 7:845–854
Kriston Á, Inzelt G (2007) J Appl Electrochem 38:415–424
Ul Hassan N, Kilic M, Okumus E, Tunaboylu B, Soydan AM (2016) J Electrochem Sci Eng 6:9–16
Sopčić S, Kraljić Roković M, Mandić Z (2012) J Electrochem Sci Eng 2:41–52
Sopčić S, Mandić Z, Kraljić Roković M (2014) Acta Chim Slov 61:272–279
Eladeb B, Bonnet C, Favre E, Lapicque F (2012) J Electrochem Sci Eng 2:211–221
Veseli A, Hajrizi A, Arbneshi T, Kalcher K (2012) J Electrochem Sci Eng 2:199–210
Velichenko AB, Luk’yanenko TV, Nikolenko NV, Amadelli R, Danilov FI (2007) Russ J Electrochem 43:118–120
Velichenko AB, Devilliers D (2007) J Fluor Chem 128:269–276
Velichenko AB, Girenko DV, Danilov FI (1995) Electrochim Acta 40:2803–2807
Chang H, Johnson DC (1989) J Electrochem Soc 136:23–27
Gilroy D, Stevens R (1980) J Appl Electrochem 10:511–525
Li LJ, Fleischmann F, Peter LM (1989) Electrochim Acta 34:459–474
Ramamurthy AC, Kuwana T (1982) J Electroanal Chem 135:243–255
Laitnen HA, Watkins NH (1976) J Electrochem Soc 123:804–809
Gonzalez-Garcia J, Gallud F, Iniesta J, Montiel V, Aldaz A, Lasia A (2000) J Electrochem Soc 147:2969–2974
Acknowledgments
The authors are indebted to Professor György Inzelt for many valuable and fruitful discussions led during our cooperation in recent years. The support of Croatian Science Foundation under the project ESUP-CAP (IP-11-2013-8825) is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Burazer, S., Sopčić, S. & Mandić, Z. Anodic deposition of lead dioxide at Nafion® covered gold electrode. J Solid State Electrochem 20, 3053–3059 (2016). https://doi.org/10.1007/s10008-016-3261-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3261-0