
Abstract
Li- and Mn-rich layered Li1.2Ni0.13Co0.13Mn0.54O2 cathode material was synthesized using sonochemical method followed by annealing at 700, 800, and 900 °C for 10 h. The material was characterized by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscopy (TEM), Raman spectroscopy, and electrochemical techniques. Its performance as a cathode material for Li-ion batteries was examined. With the sample annealed at 900 °C, an initial specific capacity of 240 mAh g−1 was obtained, which decreased to 215 mAh g−1 after 80 cycles, thus retaining about 90 % of its initial capacity. In contrast, samples annealed at lower temperatures exhibited lower capacity retention upon cycling. Thus, the final annealing temperature was found to have a significant effect on the electrochemical stability of this material in terms of capacity, average voltage, and rate capability. The advantage of this synthesis, which includes a sonochemical stage, compared with a conventional co-precipitation synthesis, was also confirmed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, there is a great interest in compounds of the general formula Li x [Mn y Ni z Co w ]O2 where x > 1, x + y + z + w = 2, and y > 0.33 as high specific capacity cathode materials for Li-ion batteries. These compounds are termed Li- and Mn-rich integrated cathode materials. The stoichiometric constraints channel the syntheses of these multi-element lithiated transition metal oxides to form a multi-phase structure in which the monoclinic Li2MnO3 compound and rhombohedral Li x MO2 (where M is a mixture of transition metals with an overall stoichiometry of unity and x ≤ 1) compound are integrated in nanodomains. The rhombohedral phase is electrochemically active, while the monoclinic phase is initially inactive. It can be activated by polarization to potentials above 4.5 V via complicated processes whose mechanisms are still being explored. After this, the entire mixture becomes electrochemically active, exhibiting reversible Li-ion insertion/de-insertion with high specific capacities that may reach 250–300 mAh g−1 [1–18]. This specific capacity is about twice that of the LiCoO2 cathodes, which are currently used in most commercial Li-ion batteries.
It follows that these cathode materials are important, having the potential to take Li-ion batteries way beyond their current level of development in terms of specific energy density. Despite their high specific capacity, these materials suffer from capacity fading and average discharge voltage decay on prolonged cycling to potentials higher than 4.6 V. Thus, stabilizing their specific capacity has become a major challenge for researchers. Several approaches such as surface coating [19–22], doping with various cations (K, Mg, Al, Fe, etc.) [17, 23–27], and the use of additives to electrolytes are reported to stabilize the specific capacity of these cathode materials [28].
Several strategic synthetic approaches have been developed to prepare Li- and Mn-rich layered cathode materials such as Li1.2Ni0.13Co0.13Mn0.54O2 (or xLi[Li1/3Mn2/3]O2.(1 − x) LiMn1/3Ni1/3Co1/3O2 (x = 0.6) for Li-ion batteries. The synthetic routes used included solid-state methods [11, 12], resorcinol formaldehyde gel [13], carbonate co-precipitation [14], sol-gel [15], spray drying [16], hydrothermal [17], and aerogel template [18] methods. The physico-chemical properties such as particle size, morphology, and porosity, etc. of materials are influenced by the synthesis method, which in turn affect the electrochemical performance of the products significantly. Generally, the lithiated metal oxides are prepared by high-temperature calcinations of precursor samples. The integration of both phases of Li2MnO3 and Li x MO2 at nanodomain in the precursor is essential in order to exhibit better electrochemical performance of these Li- and Mn-rich cathodes. The synthesis method indeed plays an important role in the electrochemical performance of these cathodes [15], where a higher specific capacity has been reported for Li1.2Ni0.13Co0.13Mn0.54O2 synthesized by self-combustion reaction as compared to sol-gel and co-precipitation method.
In recent years, sonochemical processes have become widespread to generate materials in solution media in which molecules undergo a chemical reaction due to the application of powerful ultrasound radiation (20 kHz to 1 MHz) [29]. The sonochemical synthetic methods have been effectively applied to prepare different nanomaterials such as composite materials [30], metals and alloys [31], and metal oxides [29, 32]. The physiochemical phenomenon responsible for the sonochemical processes is acoustic cavitation, which includes formation, growth, and finally implosive collapse of bubbles formed due to sonic radiation in liquid phases, generating unusual chemical and physical environments. The implosive collapse of sonically formed bubbles generates localized hotspots through their adiabatic compression within the gas phase of the collapsing bubbles. The conditions prevailing in these hotspots have been experimentally determined and were found to exhibit extremely high temperatures above 5000 K and pressures greater than 20 MP. In addition, successive heating and cooling rates of the order of 1010 K s−1 were obtained [29, 33]. Many attempts have been made to prepare Li1.2Ni0.13Co0.13Mn0.54O2 because this material possesses a very high specific capacity close to 250 mAh g−1. As a follow-up to these efforts, we propose here a new simple, cost-effective, efficient, and convenient sonochemical synthesis approach for preparing Li1.2Ni0.13Co0.13Mn0.54O2 cathode material. Significant differences between the precursors produced by the sonochemical method and the conventional co-precipitation method are observed, which affect the electrochemical performance of these Li- and Mn-rich cathodes. Although the effect of annealing temperatures on the stability of specific capacity and rate capability has been reported [8], but their effect on the stability of the average discharge voltage upon cycling has not yet been reported.
In the present work, we have attempted to improve the electrochemical performance of Li- and Mn-rich cathodes by performing a sonochemical stage before annealing the precursors at high temperatures (700, 800, and 900 °C) instead of surface coating or doping these cathodes. The sonochemical synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 has never been reported, to the best of our knowledge. The products were tested as a cathode material for Li-ion battery systems. The effect of the final annealing temperatures on the electrochemical performance such as specific capacity and average discharge voltage of the products was examined. Composite electrodes comprising the Li1.2Ni0.13Co0.13Mn0.54O2 material synthesized in this work exhibited a specific capacity of about 240 mAh g−1 when cycled at a C/10 rate. The materials that were finally treated at 900 °C were found to exhibit the best electrochemical performance. It became clear that cathode materials whose synthesis included a sonochemical stage were superior to materials synthesized by a conventional co-precipitation method, annealed at the same temperatures and tested in similar experiments. As similar reactants and annealing temperatures are used in both synthetic processes, the difference in electrochemical properties between the two samples can result from the application of a sonochemical stage during the synthesis of the precursors.
Materials and methods
Materials
LiOH.H2O (LiOH, 56 %; Acros Organics), C4H6CoO4.4H2O (Co 24 %; Alfa Aesar), Mn(CH3COO)2.4H2O (99 + %; Sigma-Aldrich), and Ni(CH3COO)2.4H2O (98 %; Sigma-Aldrich) were of analytical grade and were used as received.
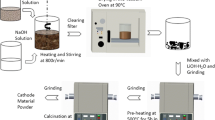
Synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 by sonochemical method
Li- and Mn-rich layered Li1.2Ni0.13Co0.13Mn0.54O2 powder was synthesized by sonochemical process combined with high-temperature calcination. In a typical process, LiOH (2.517 g, 0.3 M), Ni(CH3COO)2 (1.618 g, 0.0325 M), C4H6CoO4 (1.619 g, 0.0325 M), and Mn(CH3COO)2 (6.617 g, 0.135 M) were dissolved in 200 mL of double-distilled (DD) water (Li/Ni/Co/Mn = 1.2:0.13:0.13:0.54 in the molar ratio). The solid-liquid dispersion was sonicated for 1 h (in temperatures 0 min = ∼26 °C and 60 min = ∼80 °C) with a high-intensity ultrasonic horn (Ti-horn, 20 kHz, 750 W at 50 % efficiency, Sonics & Materials VCX600 Sonofier). The obtained solution was kept on a hot plate at 80 °C without filtration, to let the solvent evaporate. After drying in a hot-air oven at 100 °C for 10 h, the as-prepared material was ground and preheated at 400 °C for 5 h at a heating rate of 10 °C min−1. The resulting product was ground to a fine powder. It was then divided in three and heated at a rate of 10 °C min−1 in a muffle furnace until reaching 700, 800, and 900 °C. The heated powder was further calcined in air for 10 h while retaining the high temperatures, to form Li1.2Ni0.13Co0.13Mn0.54O2. The Li1.2Ni0.13Co0.13Mn0.54O2 calcined at 700, 800, and 900 °C were denoted LiR700, LiR800, and LiR900, respectively. An excess of 5 % lithium source was added to the initial material to compensate for the lithium evaporation during the calcination at high temperature [11, 34].
Synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 by conventional co-precipitation method
In a typical process, LiOH (2.517 g, 0.3 M), Ni(CH3COO)2 (1.618 g, 0.0325 M), C4H6CoO4 (1.619 g, 0.0325 M), and Mn(CH3COO)2 (6.617 g, 0.135 M) were dissolved in 200 mL DD water (Li/Ni/Co/Mn = 1.2:0.13:0.13:0.54 in the molar ratio). The reaction was allowed to proceed for 1 h while stirring at about 80 °C. The rest of the procedure was the same as described above in the section “Synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 by sonochemical method ”. The material calcined at 900 °C was denoted as LiRC900.
Characterization of the materials
The chemical compositions (Li, Ni, Co, and Mn) of the resulting materials were determined quantitatively by inductive coupled plasma atomic emission spectrometry (ICP-AES), using the Ultima-2 analyzer spectrometer from Jobin Yvon Horiba. The samples were first dissolved in a mixture of (HCl/HNO3 = 3:1) and then diluted with DD water to determine the metal ion concentration in the solution. The structure of the Li1.2Ni0.13Co0.13Mn0.54O2 samples were analyzed using an X-ray diffractometer (Bruker D8 Advanced using CuKα radiation; λ = 1.5406 Å). Micro-Raman spectroscopy measurements were performed at room temperature using an inVia micro-Raman spectrometer from Renishaw (UK) equipped with a 514-nm laser, a CCD camera, and an optical Leica microscope. A grating with 1800 lines per millimeter was used, as well as a ×50 objective lens to focus the incident beam. Raman spectra were collected from at least 5 to 10 locations in a sample. The study of the thermal decomposition of the precursor sample was analyzed by thermogravimetric (TG) analysis, carried out on a TGA/SDTA851e analysis instrument in a temperature range of 25–900 °C, at a heating rate of 10 °C min−1, in flowing air. Differential scanning calorimetry (DSC) measurement of the as-prepared sample was performed using a NETZSCH (DSC 200F3) instrument, in a temperature range of 25–600 °C, at a heating rate of 10 °C min−1, in flowing air. The powder Brunauer-Emmett-Teller (BET) specific surface area (SSA) was determined from nitrogen (N2) adsorption-desorption isotherms measured at liquid nitrogen temperature using a Nova 3200e Quantachrome analyzer. The SSA was calculated from the linear part of the BET plot. The size, morphology, and structure of the lithium-rich powders were analyzed using a high-resolution scanning electron microscope (HRSEM, FEI, Magellan 400L, accelerating voltage 15 kV) and a high-resolution transmission electron microscope (HRTEM, JEOL JEM-2100 with a LaB6 emitter operated at 200 kV). Samples for TEM measurements were prepared by making a suspension of the particles in isopropanol, using water-bath sonication. Two small droplets were then applied on a TEM copper grid, coated with a carbon film and dried in air in a covered Petri dish before sample processing.
Preparation of electrodes
The electrodes for the electrochemical studies were prepared by making slurry of 80 wt% active material, 10 wt% conductive additive super P carbon, and 10 wt% PVdF binder in N-methyl-2-pyrrolidinone (NMP) as the solvent. The slurry was uniformly coated using a doctor blade on Al foil current collectors and dried at 80 °C for 12 h in an oven. The coated Al foil was then pressed uniformly and cut into circular electrodes 14 mm in diameter. Lastly, the electrodes were dried at 110 °C for 12 h in vacuum.
Electrochemical measurements
The electrodes were tested using coin-type cells 2032 (NRC, Canada) assembled in an argon-filled dry glove box (made either by Braun or by VAC). Li metal foil was used as the counter and reference electrodes. The typical loading of the active mass was 5–6 mg cm−2. A commercial battery electrolyte solution LP 30 (Merck) consisting of 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (EC/DMC) (1:1 w/w) was used. A porous polypropylene-based membrane (Celgard) was used as the separator. The cells were stored for 12 h at room temperature after assembling, at OCV, in order to ensure the complete impregnation of the electrodes and the separators with the electrolyte solution. The cells were measured by galvanostatic charge-discharge cycling. They then underwent anodic polarization to 4.7 V, followed by cycling in the potential range of 2.0–4.6 V vs. Li/Li+ using computerized multi-channel battery testing instruments from Arbin Inc. Electrochemical impedance spectra (EIS) were recorded at various potentials with an amplitude of 5 mV around equilibrium in the frequency range of 100 kHz–0.01 Hz, using the Solartron model SI 1287 electrochemical interface and the 1255 HF Frequency Response Analyzer. Impedance data were subjected to the nonlinear least squares (NLLS) fitting procedure. The electrochemical measurements were performed at 30 °C in thermostats.
Results and discussion
The content of the metal ions and the corresponding stoichiometry of the synthesized materials were determined by ICP-AES. The molar ratio of Li/Ni/Co/Mn was found to be 1.223:0.132:0.134:0.545, 1.201:0.134:0.133:0.544, 1.200:0.130:0.130:0.543, and 1.200:0.132:0.134:0.544 for LiR700, LiR800, LiR900, and LiRC900, respectively. The materials were found to have the designated composition within a possible error of ±0.02. Based on ICP-AES results, the LiR700, LiR800, LiR900, and LiRC900 materials were found to have the following chemical formulas: Li1.223Ni0.132Co0.134Mn0.545O2, Li1.201Ni0.134Co0.133Mn0.544O2, Li1.200Ni0.130Co0.130Mn0.543O2, and Li1.200Ni0.132Co0.134Mn0.544O2, respectively. These results indicate that the excess lithium (5 %) is lost during the high-temperature calcination process [34].The concentration of Li remains almost the same once the annealing temperature equals 800 °C.
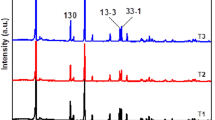
The crystal structure and the composition of the resulting materials, in their different phases, were obtained by powder XRD patterns with the Miller indices, as shown in Fig. 1a–d. The indexed peaks are consistent with the standard data found in the JCPDS files, in ICDD 2010: pdf file 01-084-1634 for the monoclinic phase and pdf file 01-075-9200 for the rhombohedral phase. The diffraction peaks are strong, sharp, and narrow, indicating the crystalline nature of the synthesized product. Moreover, no additional peaks related to impurities are observed. The obtained powder XRD peaks are indexed according to a hexagonal-layered α-NaFeO2-type structure (space group, R\( \overline{3} \)m). The presence of the monoclinic component Li2MnO3 is manifested by weak reflections in a 2θ range between 20 and 23°, which arise due to the in-plane ordering of the Li/Mn ions in the transition metal layers. These reflections originate from the ordered arrangement of the Li-ions and Ni/Co/Mn in the transition metal layers, which could be indexed by the (020) and (110) diffraction [35]. The ratio of intensities for specific XRD reflections I(003)/I(104) of sample LiR900 (1.59) is higher than those of samples LiR700 (1.46), LiR800 (1.50), and LiRC900 (1.57), which indicates improved layered structure and promising electrochemical performance.

Powder XRD patterns of Li1.2Ni0.13Co0.13Mn0.54O2 materials. a LiR700, b LiR800, c LiR900, d LiRC900, e as-prepared sample by sonochemical method, and f as-prepared sample by conventional co-precipitation method
The as-prepared sample synthesized by ultrasonic irradiation reveals high intensity, better crystallites, and sharp peaks (Fig. 1e), whereas the material obtained by a conventional co-precipitation method exhibits weak diffraction peaks, small crystallites, and is more amorphous in nature (Fig. 1f) than that obtained by the sonochemical method. Thus, the ultrasonic effect has a vital role in inducing crystallinity in the sample.
It is worth noting the differences between the LiR900 and the LiRC900 cathode materials. The XRD patterns of both materials are very similar. However, the patterns of the material prepared using ultrasonic irradiation (LiR900), after calcination, are stronger and sharper, thus showing its higher crystallinity compared with the LiRC900 material. Hence, the local high temperature and high pressure (hot spots) induced by the sonication affect not only the structure of the solid product thus formed but also, as a result, its crystallization during calcination. The higher crystallinity is beneficial to the cycling stability as well as the rate capability [36].
It is well known that sonochemistry induces very high localized temperatures compared with conventional reactions in solutions. This could be useful for the endothermic reaction that forms the mixed oxide, as was observed in Ref. [37]. Different chemical reactions can be generated by intense ultrasonic waves during ultrasonic irradiation that are strong enough to produce cavitation phenomena. These may lead to several processes such as oxidation, reduction, dissolution, decomposition, and hydrolysis [38–40]. The use of ultrasound radiation during the homogeneous precipitation of the precursors is expected to decrease the period of the precipitation reaction and to ensure fully uniform distribution of the cations within it [29].
The Raman spectra of the LiR700, LiR800, LiR900, and LiRC900 materials are compared in Fig. 2a. A very minor variation in the intensity of the Raman bands is detected upon increasing the calcination temperature and performing the synthetic procedure. The lack of pronounced differences is simply due to the fact that all the materials are very similar in their composition, phase content, and crystalline nature. Three major bands are observed in these Raman spectra with peaks at 425, 475, and 593 cm−1. They are in agreement with the theoretical predictions specified for hexagonal (R\( \overline{3} \)m) and monoclinic (C2/m) crystal structures [41]. The small peak observed at 425 cm−1 is associated with the monoclinic Li2MnO3-like short-range super-lattice ordering structure and the space group C2/m. In addition, the two other Raman peaks located at 475 and 593 cm−1 are assigned to the bending E g (symmetrical deformation) and stretching A 1g modes (symmetrical stretching of M–O), respectively [41].

a The Raman spectra of Li1.2Ni0.13Co0.13Mn0.54O2 materials; thermal analysis curves of the Li1.2Ni0.13Co0.13Mn0.54O2 precursor sample by sonochemical synthesis. b Thermogravimetric analysis (TGA) and c differential scanning calorimetry (DSC)
The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) curves of the Li1.2Ni0.13Co0.13Mn0.54O2 as-prepared sample by sonochemistry are shown in Fig. 2b, c. The thermal decomposition processes of the as-synthesized material consist of three stages. The first stage takes place in the temperature range of 25–155 °C. The calculated weight loss is 12.76 wt%, and three endothermic peaks are observed at around 85, 112, and 142 °C in the DSC curve. They are attributed to the dehydration of the lithium hydroxide, nickel hydroxide, cobalt hydroxide, and manganese hydroxide in the precursor sample. The second weight loss of 31.68 wt% is observed in the range of 155–401 °C and in the corresponding region in the DSC, reflected by a strong and sharp endothermic peak at around 289 °C. This could be ascribed to the decomposition/release of acetate moieties remaining from the cobalt, manganese, and nickel acetate precursor. The third weight loss of 10.73 wt% is detected in the temperature range of 401–560 °C, corresponding to a broad DSC exothermic peak ranging from 450 to 510 °C. This peak is attributed to the release of heat during the formation of the composite metal oxide. The calculated overall weight loss in the temperature range of 25 to 900 °C was 55.17 wt%. No weight loss was observed in the temperature range of 560–900 °C, indicating that the products’ formation is completed below 560 °C. In this temperature range, further heating leads to improvement in the crystal structure, namely, the elimination of vacancies and imperfections.
Nitrogen (N2) adsorption/desorption analysis was performed in order to investigate the BET specific surface area of the calcined materials. The BET specific surface areas of samples LiR700, LiR800, LiR900, and LiRC900 were found to be 8.3, 6.2, 3.7, and 4.1 m2 g−1, respectively, suggesting that the particle size increases with an increase in the calcination temperature. During calcination, the particle growth and partial pore fusion result in a decrease in the specific surface area. These slight differences reflected by the surface area measurements correlate well with the results obtained by SEM and TEM analyses.
The morphology and particle size of the LiR700, LiR800, LiR900, and LiRC900 materials were investigated by HRSEM and the images are shown in Fig. 3. The average sizes of the primary particles were found to be around 50–100, 80–160, 150–250, and 120–220 nm in diameter for materials LiR700, LiR800, LiR900, and LiRC900, respectively, with some agglomeration. Calcination at 900 °C seems to promote some aggregation. Further analysis by TEM and HRTEM confirmed the obtained HRSEM results (LiR700 and LiR900).

HRSEM images of Li1.2Ni0.13Co0.13Mn0.54O2 a LiR700, b LiR800, c LiR900, and d LiRC900 materials
Further studies by TEM were performed in order to examine the morphology and structure of LiR700 and LiR900 in fine detail. They included bright field (BF) imaging, conventional selected area electron diffraction (SAED) using a 600-nm aperture, and nanobeam diffraction (NBED) using a 4-nm probe. The structural assignment of the particles was based on analyzing the SAED and the NBED patterns. The information embedded in the reflections of these diffraction patterns allows in most cases to establish unambiguously the exact structure of the compounds.
Indeed, TEM provides supporting information regarding the structure of both the LiR700 and the LiR900 materials. Based on previous studies [3, 5], the results were interpreted on the basis of the two-phase system model consisting of structurally integrated layered monoclinic Li2MnO3 (C2/m) and rhombohedral LiMO2 (\( R\overline{3}m \)) components. The assigned (hkl) values in Fig. 4a–d were based on the standard ICDD 2010 data: pdf file 01-084-1634 for the monoclinic (m) phase and pdf file 01-075-9200 for the rhombohedral phase. The values are in agreement with the standard data files.

a TEM studies of Li1.2Ni0.13Co0.13 Mn0.54O2 material (LiR700); a A typical bright field image of the microstructure of the pristine material; b SAED taken with a 600 nm aperture from the particles presented in image a; c and d NBED taken with a 4-nm probe from the marked areas, showing the presence of the monoclinic (m) and the rhombohedral (rh) phases. b TEM studies Li1.2Ni0.13Co0.13Mn0.54O2 material (LiR900); a A typical bright field image of the microstructure of the pristine material; b SAED taken with a 600-nm aperture from the particles presented in image a; c and d NBED taken with a 4-nm probe from the marked areas, showing the presence of the monoclinic (m) and the rhombohedral (rh) phases. c HRTEM image displaying resolved atomic planes d = 4.75 Ǻ. The inset is the corresponding intensity profile taken from the area of the marked white rectangle. d HRTEM image displaying well-resolved atomic planes (a); b The corresponding indexed FFT from the marked area in the HRTEM image (a, the white square); c filtered and magnified image from the marked area (white square in image a).
Two modes of electron diffraction techniques:selected area electron diffraction (SAED) and nano-beam diffraction (NBED), were used to deduce the structural details from the TEM studies. The details are shown in Fig. 4a–b. SAED patterns were taken from circular areas where D = 600 nm and the convergent beam area used in the NBED technique is approximately 4 nm.
Figure 4a(a) shows a typical bright field image of a few agglomerated nanoparticles of LiR700. Submicron-sized particles of 50–100 nm are observed. The SAED (Fig. 4a(b)) image taken from these nanoparticles has a ring diffraction pattern characteristic of polycrystalline materials. The reflections that appear can be mainly indexed to the rhombohedral phase, and the presence of the monoclinic phase is suggested by the very weak reflections of the (020)m and the (110)m planes marked in Fig. 4a(b). However, probing of the local structure of nanoparticles using the NBED technique indicates the unambiguous presence of the two structurally compatible monoclinic and rhombohedral components in the LiR700. Examples of NBED patterns (Fig. 4a(c, d)), obtained from the areas marked by a white arrow (a nanoparticle) (Fig. 4a(a)), show sets of reflections (Fig. 4a(c)) which were uniquely indexed on the basis of the monoclinic unit cell. The other pattern (Fig. 4a(d)) represents the rhombohedral phase.
Mostly, HRTEM imaging has proven to be less sensitive in distinguishing between monoclinic and rhombohedral constituents because of their significant structural compatibility. As can be seen in Fig. 4c, lattice fringes with inter-planar distances of 4.75 Å, which are observed on a large number of particles, correspond either to close-packed (003) planes of LiMO2 (R\( \overline{3} \)m) layered structure or to the (001) planes of the monoclinic Li2MnO3 (C2/m) component. The measurements of the d003 or d001 were calculated as averages of 10 inter-planar distances from the line profile shown in the inset in Fig. 4c. In contrast, HRTEM imaging has helped us visualize and identify the monoclinic phase. Figure 4d shows the HRTEM image of nanoparticles revealing well-resolved crystal planes of the monoclinic Li2MnO3 phase. Based on the FFT of the partial image (white square in Fig. 4d(a)), the crystal was indexed in terms of the monoclinic unit cell of Li2MnO3. The inset in Fig. 4d(c) is the filtered image of the area that was used for the FFT (white square in Fig. 4d(a)), displaying the d(020) and d(110) families of atomic planes in the monoclinic Li2MnO3.
The TEM studies of the LiR900 material are presented in Fig. 4b. Figure 4b(a) shows a typical bright field image of a few agglomerated nanoparticles of ∼200 nm. The SAED in Fig. 4b(b) shows the presence of both the monoclinic (m) and rhombohedral (rh) phases. There are a few sets of distinctive reflections in this pattern. One set belongs to the monoclinic phase (marked with a red hexagon) and another (marked with a white hexagon) belongs to the rhombohedral (rh) phase (Fig. 4b(b)).
Using the NBED technique, we were also able to clearly show the presence of the two distinctive structures, the monoclinic and rhombohedral components. Examples of NBED patterns are presented in Fig. 4b(c, d). The patterns were obtained from the areas marked by white arrows from the nanoparticles in Fig. 4b(a). Each diffraction pattern shows sets of reflections that were uniquely indexed: one on the basis of the monoclinic unit cell (Fig. 4b(c)) and the other (Fig. 4b(d)) the rhombohedral phase.
These TEM findings show the unambiguous presence of both phases: the monoclinic and the rhombohedral. These findings are in agreement with the XRD and Raman spectra data. Also, both LiR700 and LiR900 are crystalline. The size of the obtained grains is affected by the reaction temperature: smaller grains are obtained at 700 °C and larger grains at 900 °C. The synthesis used here, with appropriate follow-up calcination, provided the expected product: a mixture at the nanometric level of crystalline rhombohedral and monoclinic phases.
Galvanostatic charge-discharge cycling experiments with cathodes comprising the materials we synthesized (LiR700, LiR800, and LiR900) were carried out vs. Li metal counter electrodes in coin cells at a C/10 rate in the potential range of 2.0–4.7 V in the first cycle followed by 2.0–4.6 V in the subsequent cycles. Typical voltage profiles thus obtained are presented in Fig. 5a–c. There are two plateaus observed during charge in the first cycle. The first plateau, below 4.4 V, corresponds to the de-intercalation of Li+ ions from the active LiNi1/3Mn1/3Co1/3O2 component. The other one, above 4.4 V, corresponds to the activation of the inactive Li2MnO3 phase, resulting in the formation of Li x MO2 (where the octahedral sites in the Li layer are partially occupied by Ni2+ cations) and Li2O [42]. The charging capacities were found to be about 300, 298, and 290 mAh g−1 for the cathode materials LiR900, LiR800, and LiR700, respectively. In the first cycle, the discharge-specific capacities were about 240, 252, and 240 mAh g−1 for LiR900, LiR800, and LiR700, respectively. The specific capacity of these Li rich metal oxide cathode materials depends on the particle size, morphology, etc., which in turn depend on the synthesis conditions. Before further discussing the electrochemical data related to this work, it is important to briefly provide some background on the performance of similar cathode materials, prepared differently by other research groups.

a–c Charge-discharge curves, d average potential vs. cycle number plots derived from galvanostatic charge-discharge curves, and (e) plots of specific capacity vs. cycle number of Li1.2Ni0.13Mn0.54Co0.13O2 electrodes at 20 mA g−1 (C/10 rate)
Zheng et al. [15] compared the electrochemical performance of Li1.2Ni0.13Co0.13Mn0.54O2 prepared by co-precipitation, sol-gel, and combustion methods and found that the sample synthesized by the combustion method exhibited a higher specific capacity than the material produced by the sol-gel and co-precipitation methods. Liu et al. [12] reported a specific capacity of 240 mAh g−1 at 30 mA g−1 for Li1.2Ni0.13Co0.13Mn0.54O2 synthesized by co-precipitation followed by a high-temperature solid-state reaction. Wang et al. [14] reported a specific capacity of 250 mAh g−1 for Li1.2Ni0.13Co0.13Mn0.54O2 synthesized by co-precipitation followed by high-temperature annealing. Shi et al. [18] compared the electrochemical performance of macroporous Li1.2Ni0.13Co0.13Mn0.54O2 synthesized by an aerogel template followed by a solid-state reaction at different temperatures and reported a value close to 240 mAh g−1 for the samples annealed at 700 and 900 °C. Hence, the values of the specific capacity measured in this work are very similar to those reported previously for Li1.2Ni0.13Co0.13Mn0.54O2 synthesized by others as described above.
Figure 5d, e provides typical cycling data of the above three cathode materials during 80 cycles. The cycling experiments were terminated after 80 cycles because too many cycles in coin-type cells reflect the influence of several components of the cells and may not provide accurate information on the intrinsic performance of the cathode materials. Likewise, comparative experiments which include several dozens of cycles provide sufficient information on the trends. Figure 5e exhibits the superiority of the cathode material annealed at 900 °C, which retains around 90 % of its initial capacity after 80 cycles, with a cycling efficiency of unity. After 80 cycles, cathodes comprising LiR800 and LiR700 materials exhibited capacity retention no more than around 71 and 63 %, respectively. Figure 5d plots the average voltage of these cathodes in terms of cycling. It is well known [9–11, 42] that the gradual decrease in the average discharge voltage of these materials is inevitable, due to the unavoidable continuous layered spinel phase transformation. The transformation occurs with all of these compounds when cycled within a wide potential domain, which includes polarization at potentials greater than 4.4 V. The plots in Fig. 5d shows that the average discharge voltage decreases during 80 cycles by 4, 4.6, and 6.6 % for LiR900, LiR800, and LiR700 cathodes, respectively. Thus, the high specific capacity and relatively high average discharge voltage of LiR900 cathodes retained during cycling show the importance of the final annealing temperature. Since the LiR900 sample exhibited sufficient improvement in electrochemical performance compared with LiR700 and LiR800, we did not increase the annealing temperature further for the preparation of the sample. Also, in a previous report, we found that when prepared at 900 °C, the material exhibited the best electrochemical performance [43]. The better electrochemical performance of this cathode material correlates with its slightly larger particle size, lower specific surface area, and higher degree of crystallinity. Figure 6 completes the general cycling experiments, showing typical voltage profiles of LiRC900 cathodes, as well as their specific capacity and average voltage after cycling compared with those of LiR900 electrodes. The discharge specific capacity was about 251 mAh g−1 for LiRC900 in the first cycle. LiRC900 electrodes exhibit similar voltage profiles to LiR900 cathodes. However, no more than 72 % capacity can be retained with the LiRC900 electrodes after 80 cycles, compared with 90 % retention of the LiR900 electrodes. The average voltage of the LiRC900 electrodes decreases by 5.6 % compared with 4 % for the LiR900 electrodes.

a Charge-discharge curves of LiRC900 electrodes at 20 mA g−1, comparison of b specific capacity and c average potential vs. cycle number from galvanostatic charge-discharge curves of LiR900 and LiRC900
In the first cycle, the discharge specific capacity was about 240, 252, 240, and 251 mAh g−1 for LiR700, LiR800, LiR900, and LiRC900, respectively. The results obtained from the material synthesized by the sonochemical and the conventional co-precipitation methods are comparable, but the capacity retention of the LiR900 cathodes is much better than that of the LiRC900 cathodes. This result is very important, indicating that the material synthesized by sonochemistry (LiR900) is superior to that produced by the conventional co-precipitation method (LiRC900). Hence, it is clear that the use of a sonochemical stage affects fine structural features, due to cavitation phenomena that induce high local temperature and pressure (hot spots). These fine structural effects lead, in turn, to better electrochemical performance of the electrodes, whose active mass preparation involved sonication. We cannot yet explain which structural changes (induced by sonication) improve the stability of these cathode materials. Though the difference in performance reported here is relatively small, the results are significant, because the improvement resulting from the use of sonochemistry is noticeable.
Rate capability was measured for both LiR700 and LiR900 electrodes. Cells containing these cathodes and Li metal anodes were cycled five times at each rate and then returned to cycling at the initial low current value as shown in Fig. 7. As expected, the specific capacity decreased with an increase in the current values for both LiR700 and LiR900 electrodes. However, there is a significant difference in the specific capacity of these types of electrodes, observed when the rate values were increased from C/2 to 2C. At a 2C rate, the specific capacities were about 90 and 125 mAh g−1 for LiR700 and LiR900 cathodes, respectively. When the rate was returned to the low current value (C/10), the high specific capacity values were again restored for both types of electrodes.

Rate capabilities of Li1.2Ni0.13Mn0.54Co0.13O2 electrodes LiR700 and LiR900 at different current values (from C/10 to 2 C rates)
Electrochemical impedance spectroscopy (EIS) is a very useful electro-analytical technique to evaluate the interfacial parameters of electrodes, including charge-transfer resistance (R ct), surface film resistance (R f), surface film capacitance (C f), and double-layer capacitance (C dl). Electrochemical impedance spectra for LiR700 and LiR900 electrodes were recorded at various equilibrium potentials with an amplitude of 5 mV in the frequency range of 100 kHz to 0.01 Hz. The cells were subjected to five galvanostatic charge-discharge cycles to activate and stabilize the electrodes before the impedance measurements. The Nyquist plots recorded at various potentials during the charge cycles are shown in Fig. 8a–d. In standard electrolyte solutions containing lithiated transition metal oxides, time constants are related to the surface films. The Nyquist plots contain a high- to medium-frequency semicircle, which is usually attributed to these time constants [9, 10]. The constants include resistance due to Li+ ion migration through the surface films (R f), coupled with film capacitance (C f). The Nyquist plots also contain a medium- to low-frequency semicircle that could be attributed to interfacial charge-transfer resistance (R ct), coupled with an interfacial capacitance (C dl). The low-frequency straight line corresponds to the solid-state diffusion of Li+ ions in the electrode, resembling Warburg-type impedance. The R ct decreases to 4.0 V and then increases after 4.2 V in the course of charging. After 4.4 V, the R ct becomes so dominant that the Warburg impedance disappears and only two semicircles are observed, in agreement with previous studies on other Li- and Mn-rich high-capacity layered cathode materials [9, 10]. When qualitatively analyzed (Fig. 8c, d), both resistance values resulting from R f and R ct were found to be greater in the case of LiR700 electrodes than they were for the LiR900 electrodes. These results correlate to the higher rate capability of LiR900 electrodes.

Nyquist plots from impedance measurements of Li1.2Ni0.13Mn0.54Co0.13O2 electrodes a LiR700 and b LiR900, qualitative comparison at c 3.8 V and d 4.4 V; e Nyquist plots of electrodes comprising LiRC900 material synthesized by a conventional co-precipitation method at various equilibrium potentials and f Nyquist plots recorded at 3.8 V during charge for LiR900 and LiRC900 electrodes, for comparison
The Nyquist plots of electrodes comprising LiRC900 was also measured at different equilibrium potentials during charge and are shown in Fig. 8e. The R ct values show a similar trend (compared to LiR900), namely, a decrease in the impedance from 3.8 V to 4.0 V and then an increase after charging beyond 4.2 V. However, both R f and R ct are found to be higher for LiRC900 than for LiR900 (Fig. 8f). The higher values of R f and R ct for LiRC900 electrodes compared with those of LiR900 electrodes calculated from EIS further reflect the inferiority of the LiRC900 material compared with the material synthesized with a sonochemical stage. The sonochemical stage seems to promote better element mixing and uniform structure of the material at the first synthesis stage. The better uniformity promotes better crystallinity and lower particle surface area and, therefore, better transport properties and lower surface reactivity.
Conclusion
In summary, for the first time, we have demonstrated a synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 which included a sonochemical stage and compared it with the conventional co-precipitation method. We then subjected it to high-temperature calcination. The cathode material LiR900 exhibited better cycling stability as well as better rate capability compared with the LiR700 and LiR900. In addition, the average discharge voltage of electrodes prepared from LiR900 was found to be always greater than that of LiR700 and LiR800 during prolonged cycling. The high specific capacity and also high discharge voltage of LiR900 resulted in a higher specific energy as compared with LiR700 and LiR800. Also, LiR900 exhibited better electrochemical performance in terms of cyclability and average voltage retention, compared with the LiRC900 synthesized by conventional co-precipitation method. LiR900 electrodes exhibited better rate capability in Li-cells compared with the LiR700 electrodes. We believe that the better performance of the LiR900 cathodes compared with all other electrodes prepared in this work is due to the higher crystallinity and uniformity and the lower surface area of the material whose synthesis included sonochemical stage and high-temperature calcination. Involving sonochemical stage in the synthesis of these materials seems to be beneficial, as the method is straightforward and cheap. Further steps in this study should include a further mechanistic investigation, which will explain how the sonochemical involvement in the synthesis really helps. The present work intends to exhibit the phenomenon and examine its extents. These cathode materials are very important due to their high specific capacity, but they involve complicated failure mechanisms. Therefore, any means that leads to their improvement (even incremental) is important and worthy of attention.
References
Thackeray MM, Johnson CS, Vaughey JT, Li N, Hackney SA (2005) Advances in manganese-oxide ‘composite’ electrodes for lithium-ion batteries. J Mater Chem 15:2257–2267
Thackeray MM, Kang SH, Johnson CS, Vaughey JT, Benedek R, Hackney SA (2007) Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J Mater Chem 17:3112–3125
Thackeray MM, Kang SH, Johnson CS, Vaughey JT, Hackney SA (2006) Comments on the structural complexity of lithium-rich Li1+x M1-x O2 electrodes (M = Mn, Ni, Co) for lithium batteries. Electrochem Comm 8:1531–1538
Yu H, Zhou H (2012) Initial Coulombic efficiency improvement of the Li1.2Mn0.567Ni0.166Co0.067O2 lithium-rich material by ruthenium substitution for manganese. J Mater Chem 22:15507–15510
Amalraj F, Kovacheva D, Talianker M, Zeiri L, Grinblat J, Leifer N, Goobes G, Markovsky B, Aurbach D (2010) Synthesis of Integrated Cathode Materials xLi2MnO3⋅ (1 − x) LiMn1/3Ni1/3Co1/3O2 (x = 0.3, 0.5, 0.7) and Studies of Their Electrochemical Behavior. J Electrochem Soc 157:A1121–A1130
Li J, Klopsch R, Stan MC, Nowak S, Kunze M, Winter M, Passerini S (2011) Synthesis and electrochemical performance of the high voltage cathode material Li[Li0.2Mn0.56Ni0.16Co0.08]O2 with improved rate capability. J Power Sources 196:4821–4825
Amalraj F, Talianker M, Markovsky B, Sharon D, Burlaka L, Shafir G, Zinigrad E, Haik O, Aurbach D, Lampert J, Dobrick MS, Garsuch A (2013) Study of the lithium-rich integrated compound xLi2MnO3·(1-x)LiMO2 (x around 0.5; M = Mn, Ni, Co; 2:2:1) and its electrochemical activity as positive electrode in lithium cells. J Electrochem Soc 160:A324–A337
Penki TR, Shanmughasundaram D, Jeyaseelan AV, Subramani AK, Munichandraiah N (2014) Polymer template assisted synthesis of porous Li1.2Mn0.53Ni0.13Co0.13O2 as a high capacity and high rate capability positive electrode material. J Electrochem Soc 161:A33–A39
Nayak PK, Grinblat J, Levi M, Markovsky B, Wu Y, Powell B, Aurbach D (2014) Structural and electrochemical evidence of layered to spinel phase transformation of Li and Mn rich layered cathode materials of the formulae xLi[Li1/3Mn2/3]O2.(1-x)LiMn1/3Ni1/3Co1/3O2 (x = 0.2, 0.4, 0.6) upon cycling. J Electrochem Soc 161:A1534–A1547
Nayak PK, Grinblat J, Levi M, Aurbach D (2014) Electrochemical and structural characterization of carbon coated Li1.2Mn0.56Ni0.16Co0.08O2 and Li1.2Mn0.6Ni0.2O2 as cathode materials for Li-ion batteries. Electrochim Acta 137:546–556
Yabuuchi N, Yoshii K, Myung ST, Nakai I, Komaba S (2011) Detailed studies of a high-capacity electrode material for rechargeable batteries Li2MnO3-LiCo1/3Ni1/3Mn1/3O2. J Am Chem Soc 133:4404–4419
Liu H, Du C, Yin G, Song B, Zuo P, Cheng X, Ma Y, Gao Y (2014) An Li-rich oxide cathode material with mosaic spinel grain and a surface coating for high performance Li-ion batteries. J Mater Chem A 2:15640–15646
Elia GA, Wang J, Bresser D, Li J, Scrosati B, Passerini S, Hassoun J (2014) A new, high energy Sn−C/Li[Li0.2Ni0.4/3Co0.4/3Mn1.6/3]O2 lithium-ion battery. ACS Appl Mater Interfaces 6:12956–12961
Wang Z, Liu E, He C, Shi C, Li J, Zhao N (2013) Effect of amorphous FePO4 coating on structure and electrochemical performance of Li1.2Ni0.13Co0.13Mn0.54O2 as cathode material for Li-ion batteries. J Power Sources 236:25–32
Zheng JM, Wu XB, Yang Y (2011) A comparison of preparation method on the electrochemical performance of cathode material Li[Li0.2Mn0.54Ni0.13Co0.13]O2 for lithium ion battery. Electrochim Acta 56:3071–3078
Yang S, Huang G, Hu S, Hou X, Huang Y, Yue M, Lei G (2014) Improved electrochemical performance of the Li1.2Ni0.13Co0.13Mn0.54O2 wired by CNT networks for lithium-ion batteries. Mater Lett 118:8–11
Li Q, Li G, Fu C, Luo D, Fan J, Li L (2014) K+-Doped Li1.2Mn0.54Co0.13Ni0.13O2: a novel cathode material with an enhanced cycling stability for lithium-ion batteries. ACS Appl Mater Interfaces 6:10330–10341
Shi SJ, Tu JP, Tang YY, Zhang YQ, Wang XL, Gu CD (2013) Preparation and characterization of macroporous Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for lithium-ion batteries via aerogel template. J Power Sources 240:140–148
Shi SJ, Tu JP, Tang YY, Liu XY, Zhang YQ, Wang XL, Gu CD (2013) Enhanced cycling stability of Li[Li0.2Mn0.54Ni0.13Co0.13]O2 by surface modification of MgO with melting impregnation method. Electrochim Acta 88:671–679
West WC, Soler J, Smart MC, Ratnakumar BV, Firdosy S, Ravi V, Anderson MS, Hrbacek J, Lee ES, Manthiram A (2011) Electrochemical behavior of layered solid solution Li2MnO3 − LiMO2 (M = Ni, Mn, Co) Li-ion cathodes with and without alumina coatings. J Electrochem Soc 158:A883–A889
Li GR, Feng X, Ding Y, Ye SH, Gao XP (2012) AlF3-coated Li(Li0.17Ni0.25Mn0.58)O2 as cathode material for Li-ion batteries. Electrochim Acta 78:308–315
Amalraj F, Talianker M, Markovsky B, Burlaka L, Leifer N, Goobes G, Erickson EM, Haik O, Grinblat J, Zinigrad E, Aurbach D, Lampert JK, Shin JY, Dobrick MS, Garsuch A (2013) Studies of Li and Mn-rich Lix[MnNiCo]O2 electrodes: electrochemical performance, structure, and the effect of the aluminum fluoride coating. J Electrochem Soc 160:A2220–A2233
Wang D, Huang Y, Huo Z, Chen L (2013) Synthesize and electrochemical characterization of Mg-doped Li-rich layered Li[Li0.2Ni0.2Mn0.6]O2 cathode material. Electrochim Acta 107:461–466
Jina X, Xua Q, Liua H, Yuana X, Xia Y (2014) Excellent rate capability of Mg doped Li[Li0.2Ni0.13Co0.13Mn0.54]O2 cathode material for lithium-ion battery. Electrochim Acta 136:19–26
Li Z, Chernova NA, Feng J, Upreti S, Omenya F, Whittingham MS (2012) Stability and rate capability of Al substituted lithium-rich high-manganese content oxide materials for Li-ion batteries. J Electrochem Soc 159:A116–A120
Lee E, Koritala R, Miller DJ, Johnson CS (2015) Aluminum and gallium substitution into 0.5Li2MnO3·0.5Li(Ni0.375Mn0.375Co0.25)O2 layered composite and the voltage fade effect. J Electrochem Soc 162:A322–A329
Nayak PK, Grinblat J, Levi M, Haik O, Levi E, Aurbach D (2015) Effect of Fe in suppressing the discharge voltage decay of high capacity Li-rich cathodes for Li-ion batteries. J Solid State Electrochem 19:2781–2792
Nayak PK, Grinblat J, Levi M, Aurbach D (2015) Understanding the effect of lithium bis(oxalato) borate (LiBOB) on the structural and electrochemical aging of Li and Mn rich high capacity Li1.2Ni0.16Mn0.56Co0.08O2 cathodes. J Electrochem Soc 162:A596–A602
Lv W, Qiu Q, Wang F, Wei S, Liu B, Luo Z (2010) Sonochemical synthesis of cobalt aluminate nanoparticles under various preparation parameters. Ultrason Sonochem 17:793–801
Malka E, Perelshtein I, Lipovsky A, Shalom Y, Naparstek L, Perkas N, Patick T, Lubart R, Nitzan Y, Banin E, Gedanken A (2013) Eradication of multi-drug resistant bacteria by a novel Zn-doped CuO nanocomposite. Small 9:4069–4076
Friedman H, Reich S, Popovitz-Biro R, von Huth P, Halevy I, Koltypin Y, Gedanken A, Porat Z (2013) Micro- and nano-spheres of low melting point metals and alloys, formed by ultrasonic cavitation. Ultrason Sonochem 20:432–444
Soltani T, Entezari MH (2013) Sono-synthesis of bismuth ferrite nanoparticles with high photocatalytic activity in degradation of Rhodamine B under solar light irradiation. Chem Engg Journal 223:145–154
Yuvaraj S, Selvan RK, Kumar VB, Perelshtein I, Gedanken A, Isakkimuthu S, Arumugam S (2014) Sonochemical synthesis, structural, magnetic and grain size dependent electrical properties of NdVO4 nanoparticles. Ultrason Sonochem 21:599–605
Sivakumar P, Nayak PK, Markovsky B, Aurbach D, Gedanken A (2015) Sonochemical synthesis of LiNi0.5Mn1.5O4 and its electrochemical performance as a cathode material for 5 V Li-ion batteries. Ultrason Sonochem 26:332–339
Xiang Y, Yin Z, Li X (2014) An improved carbonate precipitation method for the preparation of Li1.2Ni0.12Co0.12Mn0.56O2 cathode material. Ionics 20:163–168
Xue Y, Wang Z, Yu F, Zhang Y, Yin G (2014) Ethanol-assisted hydrothermal synthesis of LiNi0.5Mn1.5O4 with excellent long-term cyclability at high rate for lithium-ion batteries. J Mater Chem A 2:4185–4191
Ohayon E, Gedanken A (2010) The application of ultrasound radiation to the synthesis of nanocrystalline metal oxide in a non-aqueous solvent. Ultrason Sonochem 17:173–178
Som S, Kumar V, Kumar V, Gohain M, Pandey A, Duvenhagea MM, Terblans JJ, Bezuindenhoud BCB, Swart HC (2016) Dopant distribution and influence of sonication temperature on the pure red light emission of mixed oxide phosphor for solid state lighting. Ultrason Sonochem 28:79–89
Vijayakumar R, Koltypin Y, Felner I, Gedanken A (2000) Sonochemical synthesis and characterization of pure nanometer-sized Fe3O4 particles. Materials Science Engineering: A 286:101–105
Pinjari DV, Pandit AB (2011) Room temperature synthesis of crystalline CeO2 nanopowder: advantage of sonochemical method over conventional method. Ultrason Sonochem 18:1118–1123
Liu J, Hou M, Yi J, Guo S, Wang C, Xia Y (2014) Improving the electrochemical performance of layered lithium-rich transition-metal oxides by controlling the structural defects. Energy Environ Sci 7:705–714
Armstrong AR, Holzapfel M, Novak P, Johnson CS, Kang SH, Thackeray MM, Bruce PG (2006) Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li[Ni0.2Li0.2Mn0.6]O2. J Am Chem Soc 128:8694–8698
Chen Y, Chen Z, Xie K (2014) Effect of annealing on the first-cycle performance and reversible capabilities of lithium-rich layered oxide cathodes. J Phys Chem C 118:11505–11511
Acknowledgments
P. Sivakumar thanks the Council for Higher Education, the State of Israel, for the PBC scholarship for outstanding postdoctoral researchers from China and India. This work was partially supported by the Israel Science Foundation-ISF, as part of the INREP project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sivakumar, P., Nayak, P.K., Grinblat, J. et al. Effect of sonochemistry: Li- and Mn-rich layered high specific capacity cathode materials for Li-ion batteries. J Solid State Electrochem 20, 1683–1695 (2016). https://doi.org/10.1007/s10008-016-3176-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3176-9