
Abstract
Over the last 40 years, electrically conductive polymers have become well established as important electrode materials. Polyanilines, polythiophenes and polypyrroles have received particular attention due to their ease of synthesis, chemical stability, mechanical robustness and the ability to tailor their properties. Electrochemical synthesis of these materials as films have proved to be a robust and simple way to realise surface layers with controlled thickness, electrical conductivity and ion transport. In the last decade, the biomedical compatibility of electrodeposited polymers has become recognised; in particular, polypyrroles have been studied extensively and can provide an effective route to pharmaceutical drug release. The factors controlling the electrodeposition of this polymer from practical electrolytes are considered in this review including electrolyte composition and operating conditions such as the temperature and electrode potential. Voltammetry and current-time behaviour are seen to be effective techniques for film characterisation during and after their formation. The degree of take-up and the rate of drug release depend greatly on the structure, composition and oxidation state of the polymer film. Specialised aspects are considered, including galvanic cells with a Mg anode, use of catalytic nanomotors or implantable biofuel cells for a self-powered drug delivery system and nanoporous surfaces and nanostructures. Following a survey of polymer and drug types, progress in this field is summarised and aspects requiring further research are highlighted.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the discovery of doped polymers as electronic conductors in the 1970s, many efforts have been made to increase their conductivity and improve their synthesis, durability and ability to be processed [1, 2]. These developments have resulted in a wide range of applications over categories listed in Fig. 1.

A timeline for the history of conducting polymers and their applications
Conducting polymers can be manufactured in two distinct ways. A conductive polymer ink can be directly applied using, inkjet, screen and micro-contact printing, probe-based deposition, roll-to-roll processes such flexographic printing, soft lithography, photolithography, dip-pen nanolithography and spin coating [3, 4]. Alternatively, the monomer can be oxidised and polymerised by chemical, electrochemical, chemical vapour, vacuum and physical vapour deposition or plasma techniques [5].
Despite the development of conducting polymers in terms of active materials and manufacturing techniques, many limitations and challenges still exist. For example, some polymers are not stable and are sensitive to certain environments. Properties such as viscosity and surface tension are key parameters, and additives are necessary to improve polymer processability and stability which can reduce the polymers conductivity specifically for polymers used for drug delivery [4]. The first attempts to use conducting polymers to store and release molecules began in the 1980s [6] when Miller et al. [7] developed the first controlled release system for dopamine neurotransmitter, which is physically adsorbed and cleave-bonded to the conducting polymer using cyclic voltammetry (CV). In 1984, this was followed by the use of ferrocyanide and glutamate dopants in polypyrrole (PPy). Zinger et al. [8] were the first to report the possibility of using repetitive electrical pulses to trigger controllable small amounts of ferrocyanide ion to be released gradually from the polymer. However, the device was impractical as the amount of incorporated molecules in the polymer was so small (ca. 3.2 × 10−8 mol cm−2).
The doping process
The conductivity of the polymers can be improved by a doping process that in simple terms consists on injecting charged species into the conjugated polymer backbone by chemical, electrochemical or interfacial methods [9]. The doping process is reversible, and polymers can return to their original state with little or no degradation. In addition, doping causes changes in the volume and porosity of the film and creates the possibility to incorporate molecules. The type of doping system depends on the synthesis method; if the polymer is obtained chemically, the charge carriers are introduced into the electronic structure of the polymers via an acid-base reaction in the presence of counter ions to maintain charge neutrality. Chemical doping is easy and efficient, but it is difficult to control the dopant level and in general, inhomogeneous or incomplete doping levels are common [10]. In the electrochemical polymer synthesis, this process occurs during the oxidation or reduction reactions of the conducting polymer in the presence of doping ions. The doping level can be controlled by the applied potential to the conducting polymer used as a working electrode making the ions diffuse in or out of the structure to compensate the charge imposed on the polymer backbone. As an example, the electrochemical doping using a \( {Li}^{+} \)(\( {BF}_4^{-} \)) electrolyte can be described by the following reactions at the working electrode surface [9]:
Oxidation (p-doping)
Reduction (n-doping)
Other less common methodologies to dope the polymer include interfacial doping [9] and photochemical doping [9, 11]. In the interfacial doping, the charge carriers are injected into the polymer from the metal without the need of an ion dopant. This type of doping is used in devices such as organic light emitting diodes (OLED). The conducting polymer is sandwiched between a cathode with a low work function metal such as aluminium, to match the polymer LUMO, and an anode with a high work function metal that matches the polymer HOMO. For the photochemical doping, the polymers are exposed to the electromagnetic spectra with higher energy photons than the polymer bandgap. This excites the HOMO electrons to the conducting band. The promoted electrons produce mobile carrier charges, holes in the π-band and electrons in the \( {\uppi}^{*} \)-band, when they return to the LUMO energy level. This type of doping is used in polymers for photovoltaic devices and does not involve dopant ions [9, 11]. The following example illustrates the doping mechanism [9]:
Synthesis of conducting polymers
Of all the methodologies to obtain a doped conducting polymer, the electrochemical method is the most suitable to produce films on diverse substrates. If these substrates could be implanted in the organism, the conductive polymer can be used as drug delivery systems providing pharmaceutical products. In the case of the polymerisation of pyrrole, which has a conductivity of 18–160 S cm−1, comparable to 89–210 S cm−1 for PEDOT, the desired properties for a drug delivery system are a suitable structure and suficient homogeneity [12]. Other desired properties are briefly considered below.
Temperature
Higher temperatures increase the interaction between the monomers themselves and the formed film and also activate unwanted reactions increasing the possibility of α-β and β-β coupling instead of a free polymer defect-chain via α-α bonding. There is an inverse relationship between the temperature and the surface roughness; when the temperature decreases, the film’s surface gradually changes from rough to smooth becoming more compact, coherent and mechanically stronger. This relationship is also applicable to the conjugation chain length where a long conjugation chain with a less defective structure is formed at lower temperatures.
The doping level and the conductivity increase as the synthesis temperature decreases. However, a relatively low temperature during the synthesis might require higher electrochemical polymerisation potential. In addition, a solution containing in 0.1 M of pyrrole monomer, 0.1 M of tetrabutylammonium hexafluorophosphate (TBA-PF6) and 1 vol.% of water in propylene carbonate (PC), the conductivity of the polymer decreases when the synthesis is carried out below −40 °C [13]. This behaviour is opposite to other polymers such as polyaniline which conductivity increases when the electropolymerization was carried out in a non-aqueous solution of 1,2-dichloroethane (DCE) without a protic acid at temperatures varying from 248 to 298 K [14]. The morphology of the films deposited at low temperature is smoother and denser with smaller and more uniform grains than those prepared at higher temperatures.
pH
The pH of the electrolyte during the polymerisation of PPy influences the properties of the final film, the nature of the doping anions and the substrate material used. At low pH levels, a smooth good quality film without cracks is produced while in an alkaline medium, the film is brittle and non-uniform. In acid, the electrode potential during the polymerisation is lower than that at high pH levels and the polymerisation might coincide with the decomposition of the electrolyte. The rate of polymerisation at constant potential in the presence of the dodecylbenzenesulfonate (DBS) anion increases when the pH of the electrolyte increases from 3.2 to 10 but the PPy deposition does not occur at pH ≥ 11 even if low electrode potentials are used such as 0.65 V vs. SCE [15].
The irreversible over-oxidation potential decreases at high pH levels, and at these alkaline conditions, the electrode potential for oxygen evolution also decreases. The evolving oxygen may be the cause of the over-oxidation of the PPy film because it might destroy the conjugated structure of the polymer. In metals where oxygen evolution is prevented by a passive oxide layer, the over-oxidation of PPy was independent of the pH of the electrolyte.
Bhattacharya et al. [16] reported that the conductivity of PPy films doped with vinyl sulphonate (PPy-v) and synthesised in an acidic medium was higher (13 S cm−1) than in films synthesised in a neutral or high pH solution (6.2 S cm−1). This was caused by the presence of a carboxyl group which reacts with the oligomer radical reducing the length of the conjugated polymers. The migration of the dopant ions within the polymer instead of OH ions was cited as a possible cause of low conductivity. The authors also found that the conductivity of PPy films doped with styrene sulfonate (PPy-st) was higher than that of the PPy-v films and was not affected by the increased pH of the solution.
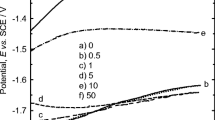
Saidman et al. [17] studied PPy growth in nitrate solutions on aluminium substrates at different pH’s. Aluminium presented a passive oxide layer which protects and inhibits the oxidation of the monomer and thus film deposition. As the pH increases, the passive layer dissolves partially allowing PPy deposition; however, the polymerisation competes with the dissolution of the metal which can prevent the deposition [17, 18]. The authors reported a homogeneous and adherent PPy film which covered the substrate surface when they applied a constant potential between 0.7 to 1.15 V vs. SCE at pH 12. They did not observe any growth of the PPy on the substrate at a pH between 4 and 11 or ≥13 [17].
Monomer concentration
Girault et al. [19] reported that the peak potential for pyrrole oxidation shifted positively by 100 mV, when the concentration of Py increased from 0.01 to 0.04 mol dm−3. They found that at 0.045 M and up to a potential of 2.1 V vs. SCE, the oxidation current rose but no oxidation peak was observed, thereby indicating the possibility of an unlimited growth of the polymer film. Iroh et al. [20] reported that the electropolymerisation rate increases with the Py concentration but the potential decreased exponentially with the monomer concentration according to the following equation:
where E p and E ox are the oxidation potentials at high and low concentration, respectively, and M is the concentration of pyrrole [21, 22]. Increasing the monomer concentration from 0.1 to 0.8 M caused a 12 % decrease in the electropolymerisation potential [22, 23]. This feature can be an advantage when a different molecule, that is electroactive at the same potential as the oxidation of Py, is incorporated into the polymer structure. Therefore, the polymerisation will occur at lower potentials than the oxidation potential of the incorporated drug molecules. This might have applications in drug delivery systems and enzymatic biosensors.
Electrolyte solution
Conducting polymers have been synthesised in ionic liquids [24], aqueous and non-aqueous solutions [25]. The nature of the electrolyte has a considerable effect on the morphology and the electrochemical and physical properties of the films [25]. For example, better quality and higher conductivity can often be obtained when the films are prepared in non-aqueous medium compared with an aqueous solution [25]. Kupila et al. [26] showed that the polymerisation efficiency and the conductance of PPy film prepared in propylene carbonate exceed that of those prepared in acetonitrile when a perchlorate counter ion was used in both systems. This may be the result of higher solubility of the dimer and oligomer species in propylene carbonate compared with their solubility in acetonitrile. Water content also has considerable effect on the film, for example, the film adherence seems to increase at low volumetric percentage of water (1–2 %). Low water content in acetonitrile led to improved PPy conductivity and low formation of the partial variation of conjugated PPy. This is because water is a stronger acid than the Py monomers and it reacts with the protons released during the polymerisation reaction hindering extra protonation of Py. Only 1 % vol. of water in acetonitrile has been proving to be optimal and sufficient to release protons during the reaction, while 1–2 % vol. improves the mechanical properties and adherence of the film [19]. In contrast, adding water to propylene carbonate does not improve the film properties and anhydrous solvents are preferred [26]. Acetonitrile is toxic and traces must be removed before any medical application; the solvent is not attractive for industrial processing due to health and safety problems.
General drug delivery systems
The choice of drug delivery methodologies depends on the drug type and treatment requirements. Conventional routes are peroral and gastrointestinal, rectal, ocular, intravaginal, transdermal, vascular injection, nasal and pulmonary [27]. While some methods are suitable for delivering certain drugs, the same method might not be appropriate for others. Taking drugs orally is probably less expensive and more convenient particularly for patients suffering from chronic diseases. Through this route, the drugs can break down by the acid environment of the stomach and by the intestine enzymes. Drug absorption in the digestive system is difficult and most macromolecules cannot be absorbed, which limits the effectiveness of the drug before reaching its target location [27–29].
Numerous attempts have been made to improve the existing drug delivery systems. A common strategy is to encapsulate the drugs with a protective layer to withstand destructive environments. The protective layer is designed to dissolve at the targeted location increasing the absorption at certain parts of the organism. Other examples include insulin injection with a needle-less injector and constant infusion pump [29].
These traditional methods cannot provide the optimum level and ensure sustained drug release. The drug concentration in the body decreases over a period of time after been introduced [30]. Additionally, a drug delivery system should provide the drug locally, for example, making neuro-growth factors in the brain available to treat neurodegenerative conditions such as Parkinsons, Alzheimers and Huntingtons and to overcome the blood–brain barrier that prevents the drug entering the brain. It has been suggested that drug delivery systems can provide anti-inflammatory medicaments and growth factors directly into the local vicinity of the implant [31]. Another application is in bone and tissue engineering where growth factors can be locally delivered at high concentrations with precise control [32–34].
Some drugs are unstable and strongly influenced by their administration time while traditional drug delivery methods often require repeated and gradual increase of dosages with toxic effects [6, 29, 35, 36].
Controllable drug delivery systems provide advantages that outweigh those offered by traditional methods. They can deliver drugs at the effective concentrations for long periods of time, without the need to take repeated doses regularly. They can be useful for patients with chronic diseases, especially those who find it difficult to adhere to a strict regimen [6].
The objective of a drug delivery system is to provide drugs to targeted location using an intermediary system that can control the administration of the drug by chemical, electrical, electrochemical, thermal or physiological release circuits or by a combination of the above [37]. Smith and Lamprou [38] have revised the applications of polymer coatings for a variety of biomedical applications including drug delivery and highlighted the main goals as the improvement of bioavailability. They also pointed out that these systems could decrease toxicity and the side effects associated to traditional methods providing protection and preservation of the drugs until they reach their target. Nevertheless, these systems demonstrated the principle of controllable molecules release. Since then, substantial progress has been made and the following sections present a discussion of the properties that make conducting polymers suitable for drug delivery systems. This will include the types of drug delivery systems, types of drugs and the necessary conditions for the drugs to be incorporated, the release methods and how these methods have been developed. The future of polymers for use in drug delivery systems and the strategies used to increase the amount of drugs that can be loaded within the polymer will also be considered.
A drug delivery system based on conducting polymers
The polymers can be chemical or electrochemically formed from an aqueous solution containing monomers, and the drug can be incorporated during the polymerisation. Various types or number of drugs and molecules whether anionic, cationic or neutral ions into the polymer backbone can be incorporated [39]. Integrating conducting polymers with other materials and nanostructures, such as titanium and carbon nanotubes, [40, 41], can increase the surface area of the films for storage. By controlling the conditions of electropolymerisation, the surface and composition with different mechanical and electrical characteristics can be obtained. Miniaturisation of polymer devices can also be used to incorporate drugs [42].
Conducting polymers undergo a reversible redox reaction which results in ion transport in and out of the polymer bulk. Typically, a potential difference less than 1 V needs to be applied between the polymer film and the electrolyte, depending on the environmental conditions, to release or capture ions. The conductive polymers can operate in a wide range of temperatures in a liquid electrolyte or in air by employing a polymer electrolyte. It is generally accepted in the literature that polypyrrole, polythiophene, polyaniline and their derivatives have some biocompatibility with living body tissues and fluids. Test for long periods of time (90 days) in vitro and in vivo have shown little evidence of toxicity or immune problems [43–47]. It has, however, been reported in literature that the polyaniline showed some cytotoxicity during in vivo studies [48].
When the conducting polymer films oxidise, its positive charge is associated with the counter ion movement from the solution into the polymer in order to compensate for the charge excess resulting in an increase in the film’s volume. In the reduction state, the counter ions are expelled from the film and cause the film to shrink. These properties can be used for drug delivery systems, where the drugs are incorporated into the polymer film during oxidation and released when the film is reduced [49].
Drug incorporation
The charged species incorporated into the polymeric matrix during the electropolymerisation process can be pharmaceutical products with different ionic charge. An example is the cationic drug risperidone incorporated onto PPy films doped with P-toluene sulfonate (PTS) anions using a galvanostatic method [50]. The freshly prepared film can release 1.1 ± 0.2 μg s−1 when ±0.6 V vs. Ag/AgCl at 0.5 Hz was applied. Other cationic molecules such as neurtrophine-3 (NT-3) have been incorporated in a PPy-PTS film during the galvanostatic polymerisation process, but the mechanism is not fully understood. It has been suggested that the electrostatic and hydrophobic interactions between the NT-3 and the doping anion PTS help to incorporate the positive NT-3 into the oxidised PPy film physically trapping the NT-3 molecules inside the polymer bulk and released during the expansion of the polymer [39].
It is also possible to incorporate a cationic drug after the polymerisation process, but the polymer needs to be doped with immobile anions such as polystyrene sulfonate (PSS). During the reduction of the polymer, the cationic drug is incorporated to compensate the negatively charged anions causing the polymer to swell. When the film oxidises, the incorporated cationic drug is ejected by electrostatic repulsion force and the film shrinks [51]. The actuation cycles of the film between the redox states may cause cracks and holes in the film, which may lead to an increase in the release rate of the molecules [6].
Anionic drugs can also be incorporated after the polymerisation process by doping the polymer with a small anion which could be the drug itself. Under these circumstances, the film oxidises by anion incorporation and is reduced by anion ejection. In the case of the incorporation of a relatively less mobile ion as a doping agent, both cationic and anionic drugs will be incorporated and expelled simultaneously. Figure 2 illustrates the mechanism of drug incorporation and release from a conducting polymer [6, 51].

Schematic for the incorporation and the release of a an anionic drug and b a cationic drug. The change in volume depends on the size of the incorporated molecules. After Svirskis, Travas-Sejdic, Rodgers and Garg [6]
Electrochemical quartz crystal microbalance (EQCM) can be used to understand the mechanism of the ion ingress and egress from the conducting polymer film and the effect of the pH and electrolyte nature. It is clear that it is preferable to use an electrolyte that becomes more acidic to promote the ingress of anionic drugs during the oxidation of a conducting polymer and ejecting them when the polymer is reduced at cathodic potentials. The redox reaction of a conducting polymer in a neutral electrolyte accepts cationic and anionic ions moving from and into the polymer film [52, 53]. If an alkaline solution is used, the incorporation of cationic drugs during the polymer reduction is facilitated. Although the slightly alkaline electrolyte may be preferred for the incorporation of cationic drugs, it may cause partial or complete deprotonation of the conducting polymer. The degree of deprotonation gradually increases with the alkalinity of the solution, and it is more likely for the oxidised PPy film than for the reduced one [53]. The deprotonation of PPy film decreased the number of charge carriers and conductivity because the hydroxyl ion reacts with proton in N-H and the residual electron recombines with the hole on the polymer backbone [53, 54]. In addition, it has been reported that the deprotonation/protonation process of a conducting polymer is reversible when is treated with an alkaline/acid solution. A complete recovery of the film conductivity has been reported when deprotonation occurs at pH values below 12 and the film is reprotonated in a 0.1 M HCl solution [53, 55].
Homogeneous films formed at low polymerisation potential or low current density are typically tightly compact and restricts the motion of molecules in and out the polymer [55]. Polymerisation using a relatively higher oxidation potential and current densities brings about the formation of a porous and more open structure film, which facilitates the ingress and release of drug substances. The augmentation of oxidation potential and current needs to be fully considered because the higher increase may activate an undesirable competitor reaction and cause over-oxidation of the polymer.
The above examples indicate that it is necessary to consider the nature of the dopant ions such as charge and size to facilitate the incorporation of the drug in the conducting polymer films [55].
Drug delivery
A conducting polymer release system can be classified broadly into several types, depending on the factors that influence the drug release. In the first type, the drug is released by a chemical method using a redox reagent that is thermodynamically able to reduce the oxidised conductive polymer or by increasing the pH. Theoretically, the conducting polymers can selectively sense the redox reagent in the solution; simultaneously, the redox reagent triggers the drug release from the conducting polymer and the drug will be released as a function of the concentration of the detected redox reagent where ionic exchange occurred. Pernat et al. [55] used a strong reducing agent hydrazine N2H4, at pH 12 to release ATP from the PPy film. The released amount was 70 nmol cm−2, which is ≈80 % less than that released by electrochemical stimulation from the same film even at N2H4 concentrations up to 10 M. The authors suggest that this is due to the low porosity of the film that unable the hydrazine molecule to penetrate and reduce the PPy film. Although hydrazine is used as an intermediate in pharmaceutical applications, it is toxic and unstable at room temperature [56]. Another example is dithiothreitol (DTT) which, despite being a strong reducing agent, failed to release any detectable ATP from the PPy film after been exposed for an hour. In addition, the incorporated drug can be released by decreasing the pH. This causes chemical deprotonation of the conducting polymer and the diffusion of the drug out of the film in parts of the body to be treated [55]. For example, Pernaut et al. [55] released the ATP drug anions from the PPy by treating the film with an alkaline NaOH solution. The alkaline solution deprotonated and reduced the conducting polymer, which resulted in the ejection of ATP ions and incorporation of hydrated sodium cations. However, the release rate due to the pH change is faster but releases ≈60 % less than the electrochemical stimulation [55].
In the second type of release system, the molecules are delivered by applying an external electrical potential to oxidise and/or reduce the films [57]. In general, two types of stimulation protocols can be used named step potential and cyclic potential. By applying a negative potential, the polymer film is reduced and its cationic charge is neutralised, which causes the ejection of the anionic drug by electrostatic force synchronised with the ingress of hydrated cations into the polymer bulk. This leads to the film swelling [31, 58]. However, holding the film under a negative stimulus potential for a period of time can cause the film to lose its electrical conductivity which is not always possible to recover [31]. Cycling the potential has the effect of moving hydrated ions in and out of the conductive polymer causing expansion and contraction which force drugs out of the film [31, 59].
Cyclic stimulation is potentially more effective and able to release higher amount of drug compared with the step potential; however, cyclic potential exposes the film to a physical stress as a result of swelling and contraction, causing delamination, cracks and breakdowns [31]. For example, it has been reported that the PPy film started to delaminate after 12 min when a cyclic potential stimulation was used to release neurotrophine-3 (NT-3) [39].
Despite the flexibility and release control provided by the electrochemical release method compared to the chemical one [55, 59, 60], this method still prevents its extensive use in in-vivo release systems because an external power supply is needed. Some drug delivery systems do not need external power sources and can use chemical, pH and temperature changes to release the drug, but such systems still have shortcomings. For example, the amount of drug released is lower than that released by the electrochemical method.
One of the problems with these systems is that the drug is released spontaneously and there is little or no control during the process. In order to avoid this problem, a multilayer system of conducting polymers has been proposed. The multilayer polymer films consist of freelance conducting layers each with a particular redox potential. For instance, Fig. 3 shows a schematic diagram of a bilayer conducting polymer system where the first layer of conducting polymer (CP1) is electrodeposited on the electrode surface from a solution containing the monomer and the anionic drug A−. The second layer CP2, with a higher redox potential than the CP1 layer, is electrodeposited on top of the CP1 surface. There is no spontaneous drug release from the system when this bilayer system is used. The unstimulated CP2 layer acts as a protective layer between the release medium and the CP1 layer [60, 61]. Once the bilayer system is fully reduced, the incorporated A− drug is released into the medium. In addition, the CP2 can be doped with other drugs which can make the system a dual-drug delivery system. It is worth noting that the oxidation potential of the first layer should be lower than the oxidation potential of the following layer; otherwise, the CP1 will act as an insulator before it can oxidise and preclude the electropolymerization of the second layer. In addition, the oxidation potential of the second layer should be in a range that does not cause over-oxidation of the first layer which thus can loss its conductivity. This condition is also valid for the other layers if the system consists of more than two layers [61].

Film consistent of bilayer conducting polymers for drug delivery system (Dual ions transport). After Pyo and Reynolds [61]
The most promising drug delivery system from those described above is the one that uses an electrical potential in order to reduce the polymer and expel the pharmaceutical product. There is also other novel and advanced possibilities; the following is a brief description.
A self-powered drug delivery system based on a galvanic cell
A limited number of studies have demonstrated a self-powered, controllable drug delivery system. Such systems can release a drug without the need of an external power source. This simplifies the manufacturing process and may reduce production costs. In addition, these systems do not need power wiring, which may limit the application of drug delivery systems, particularly for implant systems. Although most reported studies used self-powered systems to release dye molecules or model drugs (less than 10 studies used actual drugs), all relied on the same galvanic principle to generate the power.
There are three techniques used to prepare the self-powered drug delivery system based on a galvanic cell, which have been demonstrated in the literature. In the first technique, the CP is electrochemically deposited in a metal substrate such as titanium foil [57]. In the second technique, the polymer film attached or detached from a metal substrate, for example, Pt or Au, is connected to a separate anode electrode such as Zn- or Mg-based alloys [62]. The two electrodes are immersed in the same electrolyte or submerged in separate half-cell connected by a salt bridge. In the third technique, the CP film cathode coated with a thin layer of an active metal, such as Mg and Zn, serves as the anode [58]. The galvanic coupling between the Mg layer and the CP film provides the driving force for the drug release as shown in Fig. 4.

A self-powered drug delivery system based on a galvanic cell
All these techniques are based on galvanic coupling where the conducting polymer electrode is employed as a cathode and coupled with a metal electrode as an anode. Immediately, in the presence of electrolytes, the soluble metal electrode oxidises and begins dissolution, thereby reducing the CP, which causes the expulsion of the incorporated molecules. As an example, the electrochemical reactions in the magnesium anode and a PPy cathode are shown below. The anode reactions involve the oxidation of Mg metal to \( {\mathrm{Mg}}^{2+} \) ions together with hydrogen gas evolution [63]:
The cathode reaction involves the following conducting polymer (PPy) reduction:
where \( {D}^{-} \) denotes the anionic drug.
The data in Fig. 5 consider the effect of the galvanic coupling between various active metals and conducting polymers using different techniques on the release rates of model drugs and dyes.

Comparison of various drugs released from polypyrrole films using a galvanic cell. ATP release from a Ti foil: (white circle) coated PPy film (one side) and (black triangle) 20 °C and at (black hexagon) 37.5 °C coated with PPy film (one side) and eicosane-PLLA blend film (opposite side) [57]. Cumulative percentage of ATP release drug from PPy-CC composite film: (black square) uncoated composite, (white triangle) composite coated with a thin zinc layer and (black circle) composite coated with 500 nm Mg layer [58]. Cumulative percentage release of MB dye from PPy-CC composite film (black diamond) and (white diamond) coated with Mg thin layer. Release of dexamethasone in 0.1 M PBS solution from a PPy film: (sum sign) passive release; (white square) PPy film coupled with Mg-PLGA and (multiplication sign) PPy film coupled to Mg-PVA [62]
Ge et al. [57] used the first technique with the model drug adenosine triphosphate (ATP). The drug was incorporated into the polymer matrix during the electrochemical polymerisation of PPy by depositing the polymer on pure polished titanium foil. The system released 60 % of the ATP drug within 5 h at room temperature as shown in Fig. 5 (white circle). In addition, the authors reported that by coating the naked side of the titanium foil with a thin eicosane-poly (L-lactide) blend film, they reduced the released amount of the drug to 12 % under the same conditions as show in Fig. 5 (black triangle). The amount of the drug released increased by 74 % within the same period of time, when the temperature rose to human body level (37.5 °C) since the melting point of eicosane, at 36 °C, is close to body temperature, Fig. 5 (black hexagon). This can help to protect the system at room temperature and may provide an on-demand drug delivery system. However, there are only few examples of this technology and the majority of the studies focus on the coupling of metals like Zn and Mg which are easily oxidised and cause the reduction of the polymer with consequent expulsion of the drug molecule.
Magnesium is the most suitable metal for the application described above; however, the electropolymerisation of conducting polymers on its surface is challenging due to the competition between the fast dissolution or passivation of the Mg surface and the electrodeposition of the polymer. One solution is to deliberately passivate the metal surface prior the electropolymerisation with sodium salicylate in the electrolyte. Turhan et al. [64] used different doping materials during the electropolymerisation of pyrrole on Mg alloy AZ91D including carboxylic acid, sodium oxalate, sodium malonate and sodium salicylate. The result showed that PPy film was only formed when sodium salicylate salt was used. This may be due to the inability of the salt to inhibit metal dissolution. The PPy film showed a good corrosion resistance in Na2SO4. Sheng et al. [65] successfully electrodeposited PPy on zinc-coated Mg alloy AZ91D from an aqueous solution containing 0.5 M pyrrole monomer and 0.2 M sodium tartrate salt. The obtained films are homogeneous and strongly adhere to the substrate working as an effective corrosion protection coating. It appears that this methodology has not been investigated for drug delivery system. One of the problems with this technology is that the passivation of the magnesium surface prior or during the polymer deposition may prevent the electron transfer between the formed film and magnesium substrate. In this case, the drug diffuses naturally out of the polymer film.
Recently, Cui et al. [66] used cathodic potentials to deposit PPy on Mg, which helped to eliminate the dissolution of the metal. They obtained a homogenous PPy film covering the Mg foil as shown in Fig. 6a. The film was synthesised −1.9 V (Fig. 6b) and −2.8 V (Fig. 6c) vs. SCE for 1800 s from a solution containing 0.2 M Py, 0.5 NaO3 and 0.35 M HNO3. The SEM images show that the film exhibits a regular porous nanostructure PPy film as shown in Fig. 6b. In addition, the authors reported that the film can be synthesised at a potential between −2 and −2.8 V vs. SCE. However, the evolution of hydrogen bubbles significantly hinder the formation of PPy film at potentials lower than −2.8 V vs. SCE and irregular films were obtained at <−1.9 V vs. SCE as shown in Fig. 6c. Since this method avoids the formation of passive layer typically seen in the anodic deposition between Mg and PPy, this procedure offers an alternative formation of a self-powered drug delivery system.

Cathodic deposition of PPy on a magnesium substrate a image of electrode with the PPy deposited on the top part of the picture. SEM image of porous nanostructure of PPy synthesised at b −1.9 V vs. SCE and c −2.8 V vs. SCE for 1800 s [66]
Another possibility is to adsorb the polymer directly onto the Mg surface by a simple immersion in a conducting polymer solution. For example, Yfantis et al. [67] coated Mg foil AZ31 with a conducting polymer by immersing the foil in an emulsion of polyacrylic and PPy conducting polymers. The obtained films improved the corrosion rate of Mg by ≈70 % compared with uncoated Mg foil. The results showed that the corrosion resistance increased with the increase of PPy content in the coating film. By contrast, a low corrosion resistance was obtained when a PPy powder was added to the polyacrylic matrix instead of using a polypyyole-polyacrylic emulsion. This may have occurred due to changes in the physical properties of the coating film structure or the electrochemical reaction between the composite film and the Mg surface. Increasing the coating film thickness by increasing the polyacrylic content had a limited effect on the corrosion rate.
Moulton et al. [62] reported using the second methodology when the PPy film and the active metal are externally connected. The authors incorporated dexamethasone sodium drug (DEX) into PPy films as a dopant during the galvanostatic polymerisation onto a gold-coated electrode. The PPy/DEX electrode, coupled with the Mg alloy in phosphate buffer led to the dissolution of Mg and the reduction of the polymer matrix releasing the DEX drug. The final concentration of DEX was low (0.2 to 3.0 μg cm−2) but sufficient to be used as a treatment dosage.
When magnesium was covered with bioresorbable polymer such as poly (vinylalcohol) (PVA) and with poly (lactic-co-glycolic acid) (PLGA) biodegrable polymer, the release of DEX decreased compared with the test when Mg was uncoated because the amount of magnesium dissolved was low. The authors used a salt bridge to connect the two PPy and Mg coated half-cells. The final released level of DEX was 1.3 mg cm−2 (17 %) for Mg-PVA (multiplication sign in Fig. 5) and 0.2 mg cm−2 (2.6 %) for Mg-PLGA (white square in Fig. 5), compared to 3 mg cm−2 when uncoated Mg was used in the test [61]. It has been reported that the incorporated molecules diffuse into the solution after the electrode was soaked in the electrolyte for more than 30 min before the galvanic coupling. In order to minimise this diffusion and ensure that the dye was released due to the galvanic effect, a bilayer of the conducting polymers PPy-PSS was deposited on the main PPy phenol red layer, preventing the release of the phenol red dye for 4 h [62].
Wang et al. [68] incorporated phenol red salt (PR) into a PPy matrix during its electropolymerisation. The PPy-PR cathode was galvanically coupled with the Zn counter electrode in a sodium dodecyl sulphate (SDS) solution. The oxidisation of Zn and the reduction of the polymer matrix resulted in 55 % of phenol red salt released rapidly in 740 s. The rate of release declined with time, and 67 % of the dye was expelled in 60 min.
Jensen and Clark [69] used a chemical method to incorporate dyes from the phenol red class into PPy conducting polymers. Initially, they dissolved the dye in a water-ethanol mixture with pyridine. The mixture was incorporated into an oxidant solution of iron (III) p-toluenesulfonate (Fe (III)) PTS in butan-1-01 and printed on a range of substrates either directly using a pipette or using a Dimatic Materials Inkjet printer Model 2811 facilitated with a DMC-11610 cartridge (10 pL nominal injection volume). After the sample dried, the substrate was used as a polymerisation template where it was exposed to monomer vapour to form a conducting polymer film. In the second part of the experiment, the polymerisation inhibitor factor pyridine and the monomer were added into the dye-oxidant mixture and then they were applied to the substrate. The substrate was then heated for 20 min at 70 °C to evaporate the inhibitor factor and complete the polymerisation process. The dye molecules were successfully released in 0.1 M NaCl solution using Zn anode and the PPy containing the dye as a cathode on a paper coated with conducting polymer. The authors also incorporated dyes in PEDOT polymer but found that this polymer was not able to release the incorporated molecules [69].
Ge et al. [58] described a self-powered drug delivery system based on a PPy-cellulose (PPy-CC) composite film and have used the third method mentioned above. They incorporated the model drug ATP during the polymerisation process and then sputtered a thin layer of Mg or Zn at various thicknesses onto one side of the composite film. They reported that the drug release rate into 10 ml NaCl solution was slightly increased when the Mg layer grew from 450 to 500 μm. The drug release was higher when a Mg anode (90 %) was used instead of Zn (33 %) as shown in Fig. 5 (black circle and white triangle, respectively). This is because the Mg electrode potential is higher than that of Zn. The authors report a concentration of Mg2+ ions of ≈14 ppm that is safe for the humans.
Figure 5 also shows the comparison of the release of a cationic molecule such as methylene blue (MB) dye under galvanic conditions. The dye was absorbed on a (PPy-CC) composite film coated with thin magnesium. The curves in the figure show that in the presence of Mg (black diamond), the percentage released was 12 % whereas in its absence (white diamond) was ≈24 % over 5 h. The decrease is due to the reduction of the polymer because of the Mg corrosion, which leads to the reincorporation of the cationic MB on the polymer bulk and minimises its diffusion into the medium. This effect may be beneficial to eliminate spontaneous release and to control the cationic drug release over time.
It will take some time before these systems can be used in vivo because a detailed understanding of the toxicity of the metal anode, corrosion process and the amount of ion dissolution is required before the process can be used in an implant system [62]. Despite the attractive features of these systems, shortcomings remain. A number of attempts have been made to improve and control the release in order to increase the final drug concentration resulting in a high initial rate of release of molecules, but their final concentration in solution is low.
The use of catalytic nanomotors for self-powered drug delivery systems
The next generation of intelligent drug delivery systems is based on autonomous self-propelled nano- and micro-scale robotics that are able to catalytically convert chemical energy from their environment to mechanical energy [69–72]. These miniaturised systems can be divided, depending on their motion and actuation into two main types: micro/nano motors and micro/nano pumps [70, 71, 73].
The first catalytic nanomotors were discovered by Paxton et al. in 2004 [74]. The device consisted of two segments: Au and Pt. The bimetallic segments are each 1 μm long and 370 nm in diameter. The device used hydrogen peroxide fuel and relies on a self-electrophoresis mechanism. The fuel is oxidised to oxygen at the surface of the Pt section, thus causing the released electrons to be transferred through the metallic nanorods to the Au segments, where the hydrogen peroxide is reduced to water. This made nanorods in the two segments behave as a short-circuited electrochemical cell that provides a path for electrical current to flow. The electron transfer is compensated by the generation and consumption of protons on the surface of Pt and Au segments, respectively. The movements of positively charged ions create an electroosmotic flow on the nanorods/liquid interface and drag the electrolyte solution by viscosity forces, thus causing the movements of nanorods in the reverse direction by speeds that are up to ≈40 μm s−1 [71, 73]. In addition, other proposed catalytic nanomotors have used different propulsion methods, such as self-diffusiophoresis (spontaneous motion of dispersed particles in a fluid induced by a concentration gradient) [75] and bubble ejection [76]. Moreover, these miniature devices can be propelled and controlled by external stimulation, such as magnetic fields [77, 78], external electric fields [79], visible light [80], ultraviolet light [81] and ultrasonic energy [82, 83]. Catalytic nanomotors are able to perform sophisticated tasks; they communicate with each other and navigate autonomously in a microfluidic channel following the fuel concentration gradient and changing velocity depending on pH [70, 71, 84–88]. Sundararajan et al. [89] electrodeposited conducting polymer PPy on the side of the Au segments of Pt-Au catalytic motors. The nanomotors selectively pick up a positively charged polystyrene-amidine microsphere on the side of the negatively charged PPy segment at one end of the rods since the PPy segment has a more negative zeta potential than the metal segments. The presented nanomotors can transport the attached microsphere model cargo, and the motor translational motions decrease with the increase of microsphere radius. The nanorods start to move in a translational motion when the nanosphere radius increases to 1.65 μm. Ni segments were incorporated into the motor to add more control on the motion by using an external magnetic field. The segments sequence of the modified motors is Pt-Ni-Au-Ni-Au-PPy. Using a few-hundred-gauss electromagnetic field made it possible to control the direction of the movement and reduced the rotational diffusion of the rod, although the applied magnetic field decreases the motor linear movement.
It was demonstrated that the incorporated molecules can be released by using chemical stimuli [90] and the pH change of surrounding media [73, 91] or by using an external stimulation factor, such as UV light [92] and magnetic fields [93]. Theoretically, it could be possible to electrodeposit a PPy-containing drug on the nanomotor side that is then coated with Mg. The nanomotors transport the PPy cargo, which is the desired target, and the galvanic coupling between the Mg and the conducting polymer films releases the drug as shown in Fig. 7.

The concept of transportation and the release of an incorporated drug using nanomachines have been demonstrated by a number of studies. However, several problems and challenges remain and need to be resolved in order to use these machines in biomedical applications [71]. For example, the biocompatibility of these systems and their effect in-vivo application must be investigated [71]. Other areas of research ought to include the effect of the physiological environment on the operation and performance together with the interaction between the nanomotors and the surrounding medium such as electrostatic interaction with surrounding walls [71]. The precise speed control of the nanomotors and the provision of steady movement in the real 3D environment should be considered before these machines can be used in-vivo [94, 95]. These machines use toxic fuels such as hydrogen peroxide or hydrazine which may obstruct their use in biomedical application [71]. The use of a biological fuel such as glucose [96] or other biocompatible fuel may be a solution. The circulation path management of the nanomotors in the living body and their safe disposal is not clear, and further studies are needed.
Mano et al. [96] demonstrate a self-propelled bioelectrochemical motor powered by the glucose-oxygen reaction. The motor is made of carbon fibres and divided into three segments. A hydrophobic segment at the middle ranging between 6 and 10 mm and a hydrophilic, 1 mm anode and cathode at the end sides. The anode and the cathode are modified with bioelectrocatalyst redox polymer wired glucose oxide and redox polymer wired bilirubin oxidase, respectively, for the oxidation of fuel glucose and oxygen reduction. The catalytic oxidation of glucose at the anode induces electrons to move from the anode to the cathode side where oxygen is reduced. The electron stream is compensated by a proton stream through the solution from the anode to the cathode causing the motor movement. The hydrophobic segment caused the motor to float at the gas/air interface which reduces the drag force and allow the motor movement.
Zhang et al. [72] designed a self-propelled motor driven by a rapid polymerisation reaction of poly (2-ethyl cyanoacrylate) (PECA), which has been approved by the Federal Drug Administration for biomedical purposes. The motor consisted of hydroxide anion exchange resin beads (Amberlite IRA-400) soaked in PECA/acetone solution. One side of the beads was coated with poly (methyl methacrylate) (PMMA) to direct the propulsion and allow the motor to float in the electrolyte. When the motor floated in an ionic electrolyte, the hydroxide ions released from the beads surface trigger the PECA depolymerisation, causing the motor to move by 160 mm s−1 in 1 M NaCl solution. However, the motor speed significantly decreased to 10 mm s−1 when the experiment performed in relatively higher pH 7.4, in phosphate buffer saline. This may reduce the efficiency of the motor in-vivo. The drug can be incorporated in the motor fuel (PECA polymer) and released with the non-toxic products of the PECA depolymerisation. In addition, other researchers have proposed a fuel-free motor that requires external power force, such as an external magnetic field, which may complicate use in-vivo, to drive it. The suitability of these machines is still questionable because they are usually designed from a non-degradable material having the potential to generate toxic species such as nickel, chromium and silver ions [77, 97, 98]. Biocompatible and biodegradable materials instead of the non-degradable metals such as Pt are commonly used to fabricate these motors.
The chemical and electrochemical reaction between the nanomachine segments such as the likelihood galvanic corrosion of the segments should be understood before the use of these machines in real applications. Other aspects needed to evaluate the performance of the nanomachines include size and design, effect of biological substances and ions, the physiological environment such as temperature, pH, pressure, flow rate of the body fluid and tissue type [71].
The use of implantable biofuel cells for a self-powered drug delivery system
Biofuel cells, also known as biological fuel cells, can directly convert chemical energy to electricity by using biocatalyst redox reactions. The cell structure includes two electrodes with one coated with biological electrocatalyst material, such as proteins, enzymes or whole living cell organisms to catalyse the oxidation of the biofuels onto the anode electrode and/or catalyse the reduction at the cathode [99–101]. The majority of the enzymatic biofuel cells employ a reversible redox active electron transfer mediator to shuttle the electrons between the enzyme reactive site and the electrode. This type of fuel cell is identified as an indirect biofuel cell or mediated electron transfer (MET) [101, 102].
This energy conversion technology is a promising sustainable implantable electrical energy source to power several biomedical devices, such as pacemakers, neuromorphic circuits, artificial organs, implantable sensor and monitoring devices and drug delivery systems [103–106]. Zhou et al. [105] proved the concept of biofuel cells to power drug delivery systems. They demonstrated a biocomputing, logic-based, autonomous detection and self-powered controlled drug delivery system based on an enzymatic biofuel cell. The system, ‘sense-act-treat’, is made of a glassy carbon electrode modified with a carbon nanotube and Meldola’s blue. The cathode is Au coated with ((poly (3,4-ethylenedioxythiophene)–(PEDOT) containing acetaminophen drug (APAP) in 0.1 M PBS electrolyte (pH 7.4) containing nicotinamide adenine dinucleotide (NAD+) as the cofactor. The biomarkers for abdominal trauma lactic acid (LAC) and lactate dehydrogenase (LDH) are selected as a signal inputs. The results show that there is no drug release detected in the absence of one input signals ((LAC, LDH) = (0,0), (0,1) and (1,0)). In the presence of both biomarker signals ((LAC, LDH) = (1,1), the NADH was oxidised at the anode, which with the reduction of PEDOT at the cathode caused the drug’s release. The designed biofuel cell can produce a maximum power output density of 33.8 μW cm−2 at ≈0.40 V.
Other alternatives to increase the amount of drug contained in the polymer structure have been suggested. These strategies consist in increasing the surface area by incorporating nanoporous structures within the polymer or by nanostructuring the polymer itself. The next section explains the most common techniques used to obtain nanoporous surface structures and their advantages for drug release.
Conducting polymers utilising nanoporous surfaces and nanostructures
Although an increase in the thickness of the conducting polymer films increases the amount of drug that can be incorporated, the resistance of the films increases and the electroactivity decreases [107]. It has also been reported that thin films released a larger proportion of the incorporated drug than thicker films, although more molecules may also be released by thicker films by determining the appropriate electrode potential with or without longer release time [39, 49]. For example, Thompson et al. [39] reported using a 3.6 μm PPy/PTS/NT-3 film, to release 5.6 and 3.4 ng cm2 of a neurotrophic factor when the film was pulsed at ±0.5 mAcm2 at 5 Hz and unstimulated over 7 days, respectively. This amount is lower when compared with 8.8 and 5.3 ng cm2 from a thicker film 26 μm using the same release protocol and shows the importance of film thickness.
The efficiency of polymer films in releasing the drug is higher from the surface than from the bulk [59]. This may help to overcome the shortcomings of traditional conducting polymer films, which includes low capacity to load the drug associated with limited surface area. The amounts of the drug released from these films by electrical stimulation are low and not stable or sustainable, which leads to restricted application. Researchers are attempting to develop and manufacture materials with well-controlled structures at the nanometre scale for various applications. In the area of controlled drug delivery systems that are based on conducting polymer films, the construction of conducting polymer nanostructures is an effective way to increase the surface area of polymer films and thus increase the efficiency of the integration and release of a drug. Figure 8 compares the release of some previous studies of different drug release percentage using nanostructured PPy films.

Effect of nanostructured films for drug release of different molecules. Dexamethasone from: (white triangle) thin (27 μC cm−2) and (white down-pointing white triangle) thick (700 μC cm−2) films stimulated by CV at a sweep rate 100 mV s−1 [108]. (Black circle) Risperidone release from nanostructured PPy stimulated at ±0.6 V Ag/AgCl; 0.5 Hz and (black down-pointing black triangle) without electrical stimulation, (white square) release from conventional PPy films stimulated at ±0.6 V vs. Ag/AgCl; 0.5 Hz and (black triangle) without electrical stimulation [115]. Aspirin release: (black square) from conventional non-stimulated PPy, (white diamond) unstimulated nanostructured PPy, (black diamond) conventional stimulated PPy (black hexagon) stimulated nanostructure at −0.6 V vs. SCE [139]
Membrane porosity and thickness
Nanoporous membranes are used as a template where one side of the membrane is coated with conducting material to serve as a working electrode, and then the conducting polymer is electrodeposited into the pores of the other side. After attaining the required thickness by controlling the total charges passing through the polymerisation process, the nanoporous membrane template is dissolved with an appropriate material. A synthesis drug delivery system, designed by Leprince et al. [108], used this methodology; it was electrically controlled to deliver and release the anti-inflammatory drug dexamethasone (DEX). The procedure consisted on evaporating gold on one side of a nanoporous polycarbonate template to form a layer of 21 μm thickness as a working electrode. Then, platinum was electrochemically deposited into the pores of the other side of the polycarbonate template. After removing the polycarbonate template, the PPy/DEX was potentiostatically deposited on the resulting platinum nanopillar brush that resulted from dissolving the polycarbonate template [108].
The PPy/DEX nanostructured electrode was cycled in 20 mM PBS with 150 mM NaCl at room temperature and pH 7 between −0.8 to 0.9 V vs. Ag/AgCl by CV at 100 mV s−1. The reduction and oxidation peak of PPy/DEX film on the nanostructured electrodes appeared at −0.2 and 0.15 V vs. Ag/AgCl, respectively, which is significantly lower compared to oxidation potential peaks when planar electrodes were used. This decrease in the operating potential for the drug release is due to the increase in the surface area. The positive adhesion between the polymer and the electrode metal substrate improved the mechanical stability of the PPy film; unlike the flat PPy films, the nanostructured PPy/DEX film did not show cracks or delamination on after 150 CV stimulation cycles to release the drug suggesting that this design may improve the efficiency of the polymer electrodes in the long term [108].
Increasing the film thickness could enhance the amount of drug released without affecting the profile [8, 39, 108]. For example, Leprince et al. [108] reported the manufacture of two films that consumed 27.4 and 700 μC cm−2 during the electropolymerisation. Cyclic voltammetry stimulation of these films between −0.8 to 0.8 V vs. Ag/AgCl released 39 and 106 μg from the thin (27.4 μC cm−2) and thick (7004 μC cm−2) films, respectively, after 150 cycles as shown in Fig. 8 (white triangle and white down-pointing triangle, respectively). This amount is three times more for an increase in film thickness of 25 times which although not proportional is sufficient to alleviate the inflammatory reactions surrounding body implants. Furthermore, it has been found that the amount of drugs released from the film by diffusion without electrical stimulation was negligible.
The authors also found that the potential sweep rate affects the rate of drug released and the film properties. At high sweep rates, the drug ions inside the polymers’ bulk oscillate but they are not released as they too deep incorporated into the polymer structure and have no time to diffuse towards the solution before the cycle has changed direction. At low sweep rates, similar observations were made: if a negative potential is applied for a long time, the film becomes an electrical insulator because it loses the incorporated doping ions and the recovery of the film’s conductivity upon reverse oxidation becomes more difficult. The experiments suggest that the sweep rate should be optimised for effective drug release without losing the conductivity of the film. The authors report that 100 mV s−1 is an optimal sweep rate to release the anti-inflammatory drug dexamethasone while maintaining the characteristics of the film [108]. Jiang et al. [109] found that the sweep rate markedly affects the release of ATP drug from a PPy nanowire network coated by a PPy film considerably. The amount released increased significantly from 57 % to 89 % and 95 % when the system was stimulated at 50, 100 and 200 mV s−1, respectively, within 10 h. This suggests that the amount of drug released is not directly proportional to the thickness of the film and that the film releases molecules more efficiently from their surface rather than from the bulk of the polymer. This might have been resulted from the fact that the thicker films are less electroactive and allow lower diffusion rates of the drug molecule.
The increase of the electrode surface area enhances the incorporated drug in the PPy film but does not necessarily increase the amount of drug molecules per monomer of pyrrole. For example, Li et al. [110] used XPS to suggest that 103 and 765 pyrrole monomers units are needed to incorporate one molecule of antischistosomiasis, trichlorfon drug (TCF) in ITO and RVC electrodes coated with PPy, respectively. They reported that RVC electrode is a better system for this drug as incorporated and released larger amounts of TCF than the ITO electrodes due to its larger surface area even if more monomer pyrrole units were needed.
Closed packed colloid crystal array templates
Self-assembled polystyrene (PS) templates are used to fabricate porous materials involving several steps to obtain a uniform nanoparticle template where a conducting polymer can be electrochemically deposited [111, 112]. After deposition, the colloidal crystals can be dissolved in tetrahydrofuran, yielding porous conducting polymer films. The driving forces for the formation of close-packed (PS) crystals involve electrostatic interaction and lateral capillary force, and the ion diffusion during the evaporation is considered to be an important factor for the successful assembly of ordered structure on the latex surface [113].
Cho et al. [114] developed an electrically controlled nanoparticles release system based on nanoporous PPy conductive films which incorporated biotin (molecular probes) during the electrochemical deposition. The authors used an aqueous solution of 0.1 M Py monomers, 9 mM biotin and 0.01 M sodium dodecylbenzenesulfonate on an indium tin oxide (ITO) electrode modified with polystyrene spherical template (spherical diameter 1 μm) using a cathode potential of 0.7 V vs. Ag/AgCl. After dissolving the polystyrene template, the film was immobilised with streptavidin-coated gold (Au) NPs with a diameter of 1.4 or 5 nm at a concentration of 0.1 mg cm−3. Cyclic voltammetry from −1.0 to +1.0 V vs. SCE in 0.1 M phosphate-buffered saline (PBS) containing 5 mM mixture of K4Fe (CN)6/K3Fe (CN)6 was used to observe the electrochemical properties of the films’ surface at 50 m Vs−1. The release of biotin was stimulated between −2 and 2 V vs. Ag/AgCl for the Au-NPs for different time intervals. The results show that the porous biotinalited PPy films along with the electrical stimulation permit a controllable release of the gold nanoparticles by changing the strength of the chemical bonds between the PPy and biotin. The SEM images showed that a cleaned defined porous surface with clear spherical voids is interrelated and arranged in a similar manner to the arrangement of polystyrene pellets used in a template [114].
Luo et al. [59] designed a system to release a fluorescein, based on porous PPy. They pre-treated 3-mm-diameter GC electrodes rods by dropping 5 ml of a 1 % (w/v) PS nanobead suspension (mean diameter 46 ± 2 nm) using a micropipette. The electrodes were placed vertically until the suspension dried over several hours. The template solidified by heat treatment at 60 °C for ≈15 min. The PPy film incorporated with fluorescein was potentiostatically deposited on the GC electrode at 0.9 V vs. Ag/AgCl for 200 s in a solution containing 0.02 M Py and 0.01 M fluorescein sodium salt. Then, the polystyrene template was removed by steeping the electrode in toluene for 12 h. The film was stimulated to release the drug by applying −2 V vs. Ag/AgCl for 10 s in a cell containing 1.6 ml 0.1 M PBS (pH 7.4). The amount of released fluorescein from the porous PPy film was ≈10 times more than the released amount from the non-porous PPy film synthesised at the same conditions. This suggests that the porous surface increased the surface area of the film and, thus, increased the amount of the incorporated fluorescein promoting more effective drug release. In addition, the amount of the released fluorescein from the nanoporous film without electrical stimulation (pure diffusion) is negligible (3 %) compared to the control released using electrical stimulation. This indicates that the drug release system based on nanoporous PPy is a true controlled system [59].
The value of the negative applied potential on the amount of released drug was evaluated. It was observed that a gradual increase in the released drug amount will occur when a fixed potential between 0 and −2 V vs. Ag/AgCl is applied. It is interesting that some released amount of drug was observed when a positive potential 0.5 V vs. Ag/AgCl was applied. The release of the drug at a positive potential may have been caused by the negative capacitive current that surged at the end of the positive potential pulse or due to film actuation.
Sharma et al. [115] reported the electropolymerisation of PPy on a 3-dimensionally macroporous poly-methyl methacrylate (PMMA) colloidal crystal template of ≈430 nm diameter supported on stainless steel. A cationic drug, risperidone, was physically entrapped inside the macroporous PPy film (6–7 μm thick) by dropping 20 μL of methanol containing 0.1 M risperidone. The colloidal template PMMA was removed by chemical etching, and the drug was entrapped by electropolimerisation of a second PPy thin film (<0.5 μm) on top of the macroporous film to avoid spontaneous release. The macroporous film released a considerable amount of risperidone drug, 162.69 ± 3.6 μg under the application of ±0.6 V vs. Ag/AgCl at 0.5 Hz over 1 h (black circle in Fig. 8). When the film was not stimulated, the release over an hour was 119.8 ± 2.5 μg (black down-pointing triangle Fig. 8). This compares well with the amount released from a non-porous PPy film which, as can be seen in Fig. 8 (white square), is 42.5 ± 0.737 μg and 31.3 ± 0.4 μg for stimulated (±0.6 V vs. Ag/AgCl at 0.5 Hz) and non-stimulated films (black triangle Fig. 8), respectively. The increase of the drug release from the macrostructure can be ascribed to the higher amount of the incorporated within the high surface area of the modified film. The authors reported that the macroporus polypyrrole film has a higher surface area of 19.2 m2 g−1 compared to that of the plain polypyrrole film at 4.8 m2 g−1.
Nanotubular structures
The unique properties of carbon nanotubes (CN) such as high conductivity and surface area [116] have made them suitable for many areas of research such as: sensors [117], field effect transistors [118], biological materials [119], hydrogen storage [120], solar cells [121] and fuel cells [122]. As for medical applications, the reports on their toxicity are still debatable but the efficacy of carbon nanotubes to deliver a variety of drugs ranging from small molecules to peptides and proteins has been demonstrated.
Carbon nanotubes can have different mechanism to transport drug molecules; they can form covalent or non-covalent bonds with the chemical molecule, or they can be absorbed within their cavities. However, the surface tension of the liquid inside the nanotubes can reduce the effectiveness of the filling. In addition, once a certain drug is inside the nanotubes, the drug tends to diffuse out in an uncontrolled manner. A possible solution is to close the ends of the filled nanotubes by depositing a conducting polymer to control the drug release [40].
However, there are many reports of the toxicity caused by single-walled carbon nanotubes (SWCNT) and multiwall carbon nanotubes (MWCNT) such as oxidative stress of human keratinocyte cells [123, 124], inflammatory and fibrotic reactions in rats’ lungs [125] and inhibition of human HEK293 cells [126].
Ivanova et al. [127] synthesised an antibiotic and a virus control system based on MWCNT/polyaniline composite. The results revealed that MWCNT coated with PANI are better sorbent for influenza viruses than the carbon nanotubes alone. The viruses’ titre before sorption was 64 and was reduced to 16 after sorption in MWCNT, 8 in MWCNT/PANI and 4 in PANI base. These results indicate that the absorption of the viruses strongly depends on the absorbent materials.
The same authors investigated the sorption of the following antibiotics: Gramicidin S, Teicoplanin Az, Bleomycetin and Polymyxin B in MWCNT covered with polyaniline. A solution of 600 μl containing a concentration 0.2 mg ml−1 of the antibiotics was added to different amounts of carbon nanotubes 1.25, 2.5 and 5 mg and incubated for periods of different time: 15 min, 1 h and 18 h at 22 °C. This was followed by separation of the antibiotic solution from the absorbent (carbon and viruses) by a centrifuge at 5600–8850 rpm for 5–7 min. The amount of antibiotics in solution had been calculated and analysed before and after the absorption using reversed-phase chromatography (RP HPLC) on microcolumn liquid chromatograph using a multichrome-spectrum programme. The results showed that the hydrophobic antibodies Gramicidin S and teicoplanin A2 were removed within 1 h, while the removal of the plymyxin B and bleomycetic, the hydrophilic antibodies, took 18 h. This indicates that the absorption of antibiotics depends on the hydrophobic properties of the sorption materials and the antibiotics structures.
Metal oxide nanotubes, such as nanotubular titania (TiO2), which were discovered in the 1990s, are non-toxic and potentially useful materials for many applications, including biomedical ones [128–130] and are thermally stable and corrosion resistant [131–133]. They have the same advantages associated with scale that are found in carbon nanotubes, and unlike carbon nanotubes, they can be manufactured at low cost by hydrothermal, electrochemical and surfactant template techniques, [132, 134, 135] This makes PPy/TiO2 nanocomposites a good candidates for drug delivery systems [131]. However, TiO2 have large bandgap and can be considered as semiconductor materials [136, 137].
Noh et al. [138] designed a drug delivery system using aluminium oxide nanotubes. A model drug, amoxicillin, was loaded onto the internal nanotube structure and subsequently permitted to diffuse in a phosphate buffer saline solution (PBS) at a defined rate. The system showed a high rate of release in the first 6 h, and the highest released drug amount was 13 μg in the first hour. A relatively steady release profile was achieved after 7 days. The system demonstrated sustained drug release over 35 days, and the amount of drug released was proportional to the square root of time. However, anodic aluminium oxide is an electrical insulator and a passive system. Therefore, the drug release is only controlled by diffusion.
Other nanomaterials such as palygorskite clay which consist of fibrillar single crystals of 20–30 nm diameters can also be used to construct a nanostructure conducting polymer. For example, Kong et al. [139] used it to construct a nanocomposite film to absorb and release aspirin. The film was deposited at 0.80 V vs. SCE for 500 s, onto an indium doped tin oxide glass (ITO) working electrode from an electrolyte consisting of 0.56 g palygorskite clay, 75 mg of aspirin and 0.34 ml pyrrole in 25 ml PBS at pH 3.5. The natural nanostructure palygroskite helps to increase the specific surface area of the PPy film and enhance the drug incorporation and release due to the high specific surface area, high adsorption and good stability. Although the authors reported that the incorporation of the non-conductive clay had reduced the electrochemical activity of the film, the electrochemical effective surface area increased significantly from 0.72 to 4.04 cm2 for conventional and nanostructured films, respectively. The amount of aspirin released increased due to the large surface area but also due to other processes including doping, adsorption and ion dipole interactions between the carbonyl groups of aspirin anions and hydrated palygorskite cations (Mg2+, Al3+ and Fe3+). The result shown in Fig. 8 indicates that the aspirin released from the nanostructured polymer after 160 min increased from 720 μg (white diamond) to 1527.5 μg (black hexagon) from unstimulated and electrically stimulated films at −0.6 V vs. SCE, respectively. This is higher than the amount released from the conventional PPy film which was 320 and 870 μg when the same procedure was used (black square and black diamond, respectively).
Playgroskite with conducting polymers could enhance the incorporation and release of other drugs and eliminate the problem associated with some hard templates such as anodised aluminium oxide (AAO) and colloid frameworks where the templates need to be removed which increases the synthesis time or cause degradation of the drug.
Conclusions
-
1.
Drug delivery systems can benefit from high concentration of pyrrole monomers since this prevents the electrochemical active drugs reacting during the polymerisation.
-
2.
Low-temperature and low current densities will lead to less defective polymer, high doping levels, high conductivity and high electrochemical stability; however, the synthesis cost could be high.
-
3.
Higher currents and oxidation potentials will form a porous and more open polymer film structure, which facilitates ingress and release of drugs whereas low polymerisation potentials might produce low-quality polymer films.
-
4.
Tightly compact polymer structure can impede the motion of drug molecules to and from the conducting film. The oxidation potential and current need to be fully considered because the potential increase may activate an undesirable secondary reaction or over-oxidation of the polymer.
-
5.
Drug delivery systems based on conducting polymers have the potential to be used locally in order to provide the required concentration for long time periods of time, without the need for repeated doses at frequent intervals. These systems can lower drug toxicity and side effects, providing protection and preservation of the drugs until they reach their target, resulting in an improvement in drug absorption rates.
-
6.
The drug release is more efficient from the surface of the conducting polymer films than from the polymer bulk. By increasing the film thickness, the amount of drug released is higher, but this increase is not proportional and does not affect the release profile. It is possible that this is due to the less electroactivity and lower diffusion rates observed in thicker films.
-
7.
The spontaneous drug release can be eliminated by using multilayers of conducting polymer films with different redox potentials. The layer in contact with the electrolyte prevents spontaneous drug release. If the polymer layers are doped with other drugs, a dual-drug delivery system could be implemented.
-
8.
The application of an electrical potential to a PPy film causes the release of the drug; however, an external power source restricts its use in vivo.
-
9.
Cyclic voltammetry can be used for drug absorption and release. The amount is affected by the potential sweep rate, and the film can be exposed to physical stress as a result of swelling and de-swelling during the cyclic potential, causing cracks and delamination. The sweep rate should be optimised for effective drug release to keep the integrity of the film.
-
10.
Galvanic coupling between a biocompatible reactive anode, such as Mg and the conducting polymer film cathode could be used as an autonomous, self-powered source and controller for a long-term implant drug delivery system.
-
11.
The polymerisation of conducting polymers on a surface of reactive metals such as Mg is a difficult and challenging task due to the competition between the dissolution of the metal and the electrodeposition of the polymer. Coating Mg with a less active metal and the use of sodium salicylate salt during the polymerisation process may passivate the metal and allow polymerisation on reactive metals.
-
12.
The polymerisation potential of pyrrole on nanoporous and nanostructured electrodes is lower than on a flat electrode and provide high surface area for larger amount of drug storage.
-
13.
The next generation of intelligent drug delivery systems may be based on autonomous self-propelled nano- and micro-scale robots able to transport the conducting polymer’s drug cargo to the desired part of the body.
-
14.
The literature reports wide range of conducting polymers for drug delivery, but the amount of incorporated and released drug is not always mentioned. A considerable number of these studies express the released drugs in different units which makes difficult to compare. Often, the reports do not refer to the thickness of the film or to the exposed surface area. Some experiments report data captured over several days while others only consider a few seconds.
-
15.
There is an urgent need to reach a consensus on a protocol for conducting release experiments and reporting the results.
-
16.
Conducting polymer drug delivery systems still suffer from obstacles that prevent their extensive use. In particular, the storage capacity is limited; the amount of drug being released is very small, and the initial spontaneous drug release from the conducting polymer is high.
Future work
Considerable challenges still prevent the use of drug delivery systems in-vivo. These obstacles need to be resolved in order to allow this technology to be used. Some future considerations are outlined below:
-
1.
Intrinsic conducting polymers are usually not biodegradables and will need body surgery to extract them, increasing the risk of infection and reducing the patient’s healing and comfort. Grafting the monomers to a biodegradable side group like glycine ethyl ester could solve the problem.
-
2.
Drug release can be carried out by several methods including change of temperature, pH or electrical potential. Each separate method however has shortcomings and a more effective way could be the combination of them.
-
3.
In order to use Mg alloys as a power source for a self-powered drug delivery systems, the corrosion mechanism must be understood to control the rate of Mg corrosion in-vivo. The corrosion of Mg in vivo is a complicated process influenced by the composition and temperature of the surrounding environment. The Mg should biodegrade to provide the required power to release the drug from the conducting polymer and be expelled at the end of the treatment period. A detailed understanding of the toxicity, corrosion process and amount of ion dissolution is required before Mg can be used.
-
4.
The autonomous nanomachines face bigger challenges including biocompatible materials, operational performance, electrochemical reactions and the interaction with the surrounding media. The speed and direction in living environments need to be considered. A biocompatible fuel such as glucose should be used although a fuel-free motor powered by a magnetic field has also been proposed.
-
5.
Inert metal nanoparticles such as gold embedded in the nanostructured PPy (PPy nanowires, nanotubes) enhance the conductivity of the polymer, increase the amount of released drug and render the polymer more sensitive to an external electromagnetic field stimulator. The advantage of an electromagnetic field is that it is a non-invasive technique and provides a power source for drug release from the PPy. The use of electromagnetic field stimulator with a biodegradable nanostructured PPy is an emerging area of research [140].
References
Unsworth J, Lunn BA, Innis PC, Jin Z, Kaynak A, Booth NG (1992) Conducting polymer electronics. J Intell Mater Syst Struct 3:380–395
Wang J-Z, Chou S-L, Liu H, Wang GX, Zhong C, Chew SY, Liu HK (2009) Highly flexible and bendable free-standing thin film polymer for battery application. Mater Lett 63:2352–2354
Cho J, Shin K-H, Jang J (2010) Micropatterning of conducting polymer tracks on plasma treated flexible substrate using vapor phase polymerization-mediated inkjet printing. Synth Met 160:1119–1125
Gonzalez-Macia L, Morrin A, Smyth MR, Killard AJ (2010) Advanced printing and deposition methodologies for the fabrication of biosensors and biodevices. Analyst 135:845–867
Ummartyotin S, Wu C, Sain M, Manuspiya H (2011) Deposition of PEDOT: PSS nanoparticles as a conductive microlayer anode in OLED devices by desktop inkjet printer. J Nanomater 2011:606714
Svirskis D, Travas-Sejdic J, Rodgers A, Garg S (2010) Electrochemically controlled drug delivery based on intrinsically conducting polymers. J Control Release 146:6–15
Miller LL, Lau ANK, Miller EK (1982) Electrically stimulated release of neurotransmitters from a surface. An analog of the presynaptic terminal. J Am Chem Soc 104:5242–5244
Zinger B, Miller LL (1984) Timed release of chemicals from polypyrrole films. J Am Chem Soc 106:6861–6863
Heeger AJ (2001) Nobel Lecture: semiconducting and metallic polymers: the fourth generation of polymeric materials. Rev Mod Phys 73(3):681–700
The nobel prize in chemistry (2000) The Royal Swedish Academy of Sciences, Bengt N. http://www.nobelprize.org. Accessed 30 April 2015
Heeger AJ (2010) Semiconducting polymers: the third generation. Chem Soc Rev 39:2354–2371
Green RA, Lovell NH, Wallace GG, Poole-Warren LA (2008) Conducting polymers for neural interfaces: challenges in developing an effective long-term implant. Biomaterials 29:3393–3399
Yoon CO, Sung HK, Kim JH, Barsoukov E, Kim JH, Lee H (1999) The effect of low-temperature conditions on the electrochemical polymerization of polypyrrole films with high density, high electrical conductivity and high stability. Synth Met 99:201–212
Teshima K, Yamada K, Kobayashi N, Hirohashi R (1997) Effect of electropolymerization temperature on structural, morphological and conductive properties of poly(aniline) deposits prepared in 1,2-dichloroethane without a proton donor. J Electroanal Chem 426:97–102
Shimoda S, Smela E (1998) The effect of pH on polymerization and volume change in PPy (DBS). Electrochim Acta 44:219–238
Bhattacharya A, De A, Das S (1996) Electrochemical preparation and study of transport properties of polypyrrole doped with unsaturated organic sulfonates. Polymer 37:4375–4382
Saidman SB, Bessone JB (2002) Electrochemical preparation and characterisation of polypyrrole on aluminium in aqueous solution. J Electroanal Chem 521:87–94
Saidman SB, Quinzani OV (2004) Characterisation of polypyrrole electrosynthesised on aluminium. Electrochim Acta 50:127–134
Tietje-Girault J, Ponce de León C, Walsh FC (2007) Electrochemically deposited polypyrrole films and their characterization. Surf Coat Technol 201:6025–6034
Iroh JO, Su W (1999) Characterization of the passive inorganic interphase and polypyrrole coatings formed on steel by the aqueous electrochemical process. J Appl Polym Sci 71:2075–2086
Iroh JO, Su W (2000) Corrosion performance of polypyrrole coating applied to low carbon steel by an electrochemical process. Electrochim Acta 46:15–24
Su W, Iroh JO (1997) Formation of polypyrrole coatings onto low carbon steel by electrochemical process. J Appl Polym Sci 65:417–424
Su W, Iroh JO (1997) Formation of polypyrrole coatings on stainless steel in aqueous benzene sulfonate solution. Electrochim Acta 42:2685–2694
Kubisa P (2004) Application of ionic liquids as solvents for polymerization processes. Prog Polym Sci 29:3–12
Ko J, Rhee H, Park SM, Kim C (1990) Morphology and electrochemical properties of polypyrrole films prepared in aqueous and nonaqueous solvents. J Electrochem Soc 137:905–909
Kupila EL, Kankare J (1996) Electropolymerization of pyrrole in aqueous solvent mixtures studied by in situ conductimetry. Synth Met 82:89–95
Owens DR, Zinman B, Bolli G (2003) Alternative routes of insulin delivery. Diabet Med 20:886–898
Shaji J, Patole V (2008) Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci 70:269–277
Razzacki SZ, Thwar PK, Yang M, Ugaz VM, Burns MA (2004) Integrated microsystems for controlled drug delivery. Adv Drug Deliv Rev 56:185–198
Shaik MR, Korsapati M, Panati D (2012) Polymers in controlled drug delivery systems. Int J Pharm Sci 2:112–116
Wadhwa R, Lagenaur CF, Cui XT (2006) Electrochemically controlled release of dexamethasone from conducting polymer polypyrrole coated electrode. J Control Release 110:531–541
Venkatesan J, Bhatnagar I, Manivasagan P, Kang K-H, Kim S-K (2015) Alginate composites for bone tissue engineering: a review. Int J Biol Macromol 72:269–281
Pelto J, Björninen M, Pälli A, Talvitie E, Hyttinen J, Mannerström B, Seppanen S-R, Kellomäki M, Miettinen S, Haimi S (2013) Novel polypyrrole-coated polylactide scaffolds enhance adipose stem cell proliferation and early osteogenic differentiation. Tissue Eng 19:882–892
Shoichet M, Winn S (2000) Cell delivery to the central nervous system. Adv Drug Deliv Rev 42:81–102
Langer R (1990) New methods of drug delivery. Science 249:1527–1533
Wang P, Frazier J, Brem H (2002) Local drug delivery to the brain. Adv Drug Deliv Rev 54:987–1013
Geetha S, Rao C, Vijayan M, Trivedi DC (2006) Biosensing and drug delivery by polypyrrole. Anal Chim Acta 568:119–125
Smith J, Lamprou D (2014) Polymer coatings for biomedical applications: a review. Trans IMF 92:9–19
Thompson BC, Moulton SE, Ding J, Richardson R, Cameron A, O’Leary S, Wallace GG, Clark GM (2006) Optimising the incorporation and release of a neurotrophic factor using conducting polypyrrole. J Control Release 116:285–294
Luo X, Matranga C, Tan S, Alba N, Cui XT (2011) Carbon nanotube nanoreservior for controlled release of anti-inflammatory dexamethasone. Biomaterials 32:6316–6323
Herrasti P, Kulak AN, Bavykin DV, Ponce de Leon C, Zekonyte J, Walsh FC (2011) Electrodeposition of polypyrrole–titanate nanotube composites coatings and their corrosion resistance. Electrochim Acta 56:1323–1328
Prakash SB, Urdaneta M, Christophersen M, Smela E, Abshire P (2008) In situ electrochemical control of electroactive polymer films on a CMOS chip. Sensors Actuators B Chem 129:699–704
Smela E (2003) Conjugated polymer actuators for biomedical applications. Adv Mater 15:481–494
Ateh D, Navsaria HA, Vadgama P (2006) Polypyrrole-based conducting polymers and interactions with biological tissues. J R Soc Interface 22:741–752
Ferraz N, Strømme M, Fellström B, Pradhan S, Nyholm L, Mihranyan A (2012) In vitro and in vivo toxicity of rinsed and aged nanocellulose–polypyrrole composites. J Biomed Mater Res 100:2128–2138
Kamalesh S, Tan P, Wang J, Lee T, Kang E-T, Wang C-H (2000) Biocompatibility of electroactive polymers in tissues. J Biomed Mater Res 52:467–478
Wang X, Gu X, Yuan C, Chen S, Zhang P, Zhang T, Yao J, Chen F, Chen G (2004) Evaluation of biocompatibility of polypyrrole in vitro and in vivo. J Biomed Mater Res 68:411–422
Humpolicek P, Kasparkova V, Saha P, Stejskal J (2012) Biocompatibility of polyaniline. Synth Met 162:722–727
Kontturi K, Pentti P, Sundholm G (1998) Polypyrrole as a model membrane for drug delivery. J Electroanal Chem 453:231–238
Svirskis D, Wright BE, Travas-Sejdic J, Rodgers A, Garg S (2010) Evaluation of physical properties and performance over time of an actuating polypyrrole based drug delivery system. Sensors Actuators B Chem 151:97–102
Gandhi MR, Murray P, Spinks GM, Wallace GG (1995) Mechanism of electromechanical actuation in polypyrrole. Synth Met 73:247–256
Hepel J, Bruckenstein S, Hepel M (1997) Effect of pH on ion dynamics in composite PPy/Heparin films. Microchem J 55:179–191
Xie Q, Kuwabata S, Yoneyama H (1997) EQCM studies on polypyrrole in aqueous solutions. J Electroanal Chem 420(1–2):219–225
Inganäs O, Erlandsson R, Nylander C, Lundström I (1984) Proton modification of conducting polypyrrole. J Phys Chem Solids 45:427–432
Pernaut J-M, Reynolds JR (2000) Use of conducting electroactive polymers for drug delivery and sensing of bioactive molecules. A redox chemistry approach. J Phys Chem B 104:4080–4090
Kean T, Miller J, Skellern G, Snodin D (2006) Acceptance criteria for levels of hydrazine in substances for pharmaceutical use and analytical methods for its determination. Pharmeur Bio Sci Notes 2:23–33
Ge D, Qi R, Mu J, Ru X, Hong S, Ji S, Linkov V, Shi W (2010) A self-powered and thermally-responsive drug delivery system based on conducting polymers. Electrochem Commun 12:1087–1090
Ge D, Ru X, Hong S, Jiang S, Tu J, Wang J, Zhang A, Ji S, Linkov V, Ren B, Shi W (2010) Coating metals on cellulose–polypyrrole composites: a new route to self-powered drug delivery system. Electrochem Commun 12:1367–1370
Luo X, Cui XT (2009) Electrochemically controlled release based on nanoporous conducting polymers. Electrochem Commun 11:402–404
Luo X, Cui XT (2009) Sponge-like nanostructured conducting polymers for electrically controlled drug release. Electrochem Commun 11:1956–1959
Pyo M, Reynolds JR (1995) Poly(pyrrole adenosine 5′-triphosphate) (PP-ATP) and conducting polymer bilayers for transport of biologically active ions. Synth Met 71:2233–2236
Moulton SE, Imisides MD, Shepherd RL, Wallace GG (2008) Galvanic coupling conducting polymers to biodegradable Mg initiates autonomously powered drug release. J Mater Chem 18:3608–3613
Song Y, Shan D, Chen R, Zhang F, Han E-H (2009) Biodegradable behaviors of AZ31 magnesium alloy in simulated body fluid. Mater Sci Eng C 29:1039–1045
Turhan MC, Weiser M, Killian MS, Leitner B, Virtanen S (2011) Electrochemical polymerization and characterization of polypyrrole on Mg–Al alloy (AZ91D). Synth Met 161:360–364
Sheng N, Ohtsuka T (2012) Preparation of conducting poly-pyrrole layer on zinc coated Mg alloy of AZ91D for corrosion protection. Prog Org Coat 75:59–64
Cui X, Huang X, He Y, Dai L, Wang S, Sun Y, Shi W, Ge D (2014) Cathodic protection: a new strategy to enable the formation of nanostructured polypyrrole on magnesium. Synth Met 195:97–101
Yfantis A, Paloumpa I, Schmeißer D, Yfantis D (2002) Novel corrosion-resistant films for Mg alloys. Surf Coat Technol 151:400–404
Wang CY, Ashraf S, Too CO, Wallace GG (2009) Ionic liquid as electrolyte in a self-powered controlled release system. Sensors Actuators B Chem 141:452–457
Winther-Jensen B, Clark NB (2008) Controlled release of dyes from chemically polymerised conducting polymers. React Funct Polym 68:742–750
Patra D, Sengupta S, Duan W, Zhang H, Pavlick R, Sen A (2013) Intelligent, self-powered, drug delivery systems. Nanoscale 5:1273–1283
Pumera M (2010) Electrochemically powered self-propelled electrophoretic nanosubmarines. Nanoscale 2:1643–1649
Zhang H, Duan W, Liu L, Sen A (2013) Depolymerization-powered autonomous motors using biocompatible fuel. J Am Chem Soc 135:15734–15737
Wang W, Duan W, Sen A, Mallouk TE (2013) Catalytically powered dynamic assembly of rod-shaped nanomotors and passive tracer particles. Proc Natl Acad Sci U S A 110:17744–17749
Paxton WF, Kistler KC, Olmeda CC, Sen A, Angelo SK St., Cao Y, Mallouk TE, Lammert PE, Crespi VH (2004) Catalytic nanomotors: autonomous movement of striped nanorods. J Am Chem Soc 126:13424–13431.
Golestanian R, Liverpool TB, Ajdari A (2005) Propulsion of a molecular machine by asymmetric distribution of reaction products. Phys Rev Lett 94:220801-1–220801-4
Solovev AA, Mei Y, Bermúdez Ureña E, Huang G, Schmidt OG (2009) Catalytic microtubular jet engines self-propelled by accumulated gas bubbles. Small 5:1688–1692
Ghosh A, Fischer P (2009) Controlled propulsion of artificial magnetic nanostructured propellers. Nano Lett 9:2243–2245
Gao W, Sattayasamitsathit S, Manesh KM, Weihs D, Wang J (2010) Magnetically powered flexible metal nanowire motors. J Am Chem Soc 132:14403–14405
Loget G, Kuhn A (2010) Propulsion of microobjects by dynamic bipolar self-regeneration. J Am Chem Soc 132:15918–15919
Solovev AA, Smith EJ, Bof ’Bufon CC, Sanchez S, Schmidt OG (2011) Light-controlled propulsion of catalytic microengines. Angew Chem Int Ed 50:10875–10878
Hong Y, Diaz M, Córdova-Figueroa UM, Sen A (2010) Light-driven titanium-dioxide-based reversible microfireworks and micromotor/micropump systems. Adv Funct Mater 20:1568–1576
Wang W, Castro LA, Hoyos M, Mallouk TE (2012) Autonomous motion of metallic microrods propelled by ultrasound. ACS Nano 6:6122–6132
Garcia-Gradilla V, Orozco J, Sattayasamitsathit S, Soto F, Kuralay F, Pourazary A, Katzenberg A, Gao W, Shen Y, Wang J (2013) Functionalized ultrasound-propelled magnetically guided nanomotors: toward practical biomedical applications. ACS Nano 7:9232–9240
Pumera M (2011) Nanomaterials meet microfluidics. Chem Commun 47:5671–5680
Hong Y, Blackman NMK, Kopp ND, Sen A, Velegol D (2007) Chemotaxis of nonbiological colloidal rods. Phys Rev Lett 99:178103-1–178103-4
Wang Y, S-t F, Byun Y-M, Lammert PE, Crespi VH, Sen A, Mallouk TE (2009) Dynamic interactions between fast microscale rotors. J Am Chem Soc 131:9926–9927
Kanti Dey K, Ranjan Panda B, Paul A, Basu S, Chattopadhyay A (2010) Catalytic gold nanoparticle driven pH specific chemical locomotion. J Colloid Interface Sci 348:335–341
Dey KK, Bhandari S, Bandyopadhyay D, Basu S, Chattopadhyay A (2013) The pH taxis of an intelligent catalytic microbot. Small 9:1916–1920
Sundararajan S, Lammert PE, Zudans AW, Crespi VH, Sen A (2008) Catalytic motors for transport of colloidal cargo. Nano Lett 8:1271–1276
Orozco J, Campuzano S, Kagan D, Zhou M, Gao W, Wang J (2011) Dynamic isolation and unloading of target proteins by aptamer-modified microtransporters. Anal Chem 83:7962–7969
Campuzano S, Orozco J, Kagan D, Guix M, Gao W, Sattayasamitsathit S, Claussen JC, Merkoçi A, Wang J (2011) Bacterial isolation by lectin-modified microengines. Nano Lett 12:396–401
Sundararajan S, Sengupta S, Ibele ME, Sen A (2010) Drop-off of colloidal cargo transported by catalytic Pt–Au nanomotors via photochemical stimuli. Small 6:1479–1482
Burdick J, Laocharoensuk R, Wheat PM, Posner JD, Wang J (2008) Synthetic nanomotors in microchannel networks: directional microchip motion and controlled manipulation of cargo. J Am Chem Soc 130:8164–8165
Wang J, Manesh KM (2010) Motion control at the nanoscale. Small 6:338–345
Mirkovic T, Zacharia NS, Scholes GD, Ozin GA (2010) Nanolocomotion-catalytic nanomotors and nanorotors. Small 6:159–167
Mano N, Heller A (2005) Bioelectrochemical propulsion. J Am Chem Soc 127:11574–11575
Zhang L, Abbott JJ, Dong L, Kratochvil BE, Bell D, Nelson BJ (2009) Artificial bacterial flagella: fabrication and magnetic control. Appl Phys Lett 94:064107
Gao W, Kagan D, Pak OS, Clawson C, Campuzano S, Chuluun-Erdene E, Shipton E, Fullerton EE, Zhang L, Lauga E, Wang J (2012) Cargo-towing fuel-free magnetic nanoswimmers for targeted drug delivery. Small 8:460–467
Palmore GTR, Whitesides GM (1994) Microbial and enzymatic biofuel cells. ACS Symp Ser 566:271–290
Osman MH, Shah AA, Walsh FC (2010) Recent progress and continuing challenges in bio-fuel cells. Part II: microbial. Biosens Bioelectron 26:953–963
Neto SA, De Andrade AR (2013) New energy sources: the enzymatic biofuel cell. J Braz Chem Soc 24:1891–1912
Osman MH, Shah AA, Walsh FC (2011) Recent progress and continuing challenges in bio-fuel cells. Part I: enzymatic cells. Biosens Bioelectron 26:3087–3102
Oliver NS, Toumazou C, Cass AEG, Johnston DG (2009) Glucose sensors: a review of current and emerging technology. Diabet Med 26:197–210
Zhou M, Du Y, Chen C, Li B, Wen D, Dong S, Wang E (2010) Aptamer-controlled biofuel cells in logic systems and used as self-powered and intelligent logic aptasensors. J Am Chem Soc 132:2172–2174
Zhou M, Zhou N, Kuralay F, Windmiller JR, Parkhomovsky S, Valdés-Ramírez G, Katz E, Wang J (2012) A self-powered “sense-act-treat” system that is based on a biofuel cell and controlled by Boolean logic. Angew Chem Int Ed 51:2686–2689
Bullen RA, Arnot TC, Lakeman JB, Walsh FC (2006) Biofuel cells and their development. Biosens Bioelectron 21:2015–2045
Li L, Huang C (2007) Electrochemical/electrospray mass spectrometric studies of electrochemically stimulated ATP release from PP/ATP films. J Am Soc Mass Spectrom 18:919–926
Leprince L, Dogimont A, Magnin D, Demoustier-Champagne S (2010) Dexamethasone electrically controlled release from polypyrrole-coated nanostructured electrodes. J Mater Sci Mater Med 21:925–930
Jiang S, Sun Y, Cui X, Huang X, He Y, Ji S, Shi W, Ge D (2013) Enhanced drug loading capacity of polypyrrole nanowire network for controlled drug release. Synth Met 163:19–23
Li Y, Ewen RJ, Campbell SA, Smith JR (2012) Electrochemically controlled release of antischistosomiasis agents from polypyrrole. J Mater Chem 22:2687–2694
Liu Z, Ya J, Xin Y, Ma J, Zhou C (2006) Assembly of polystyrene colloidal crystal templates by a dip-drawing method. J Cryst Growth 297:223–227
Li S, Zheng J, Zhao Y, Liu Y (2008) Preparation of a three-dimensional ordered macroporous titanium dioxide material with polystyrene colloid crystal as a template. J Appl Polym Sci 107:3903–3908
Zeng F, Sun Z, Wang C, Ren B, Liu X, Tong Z (2002) Fabrication of inverse opal via ordered highly charged colloidal spheres. Langmuir 18:9116–9120
Cho Y, Borgens RB (2011) Biotin-doped porous polypyrrole films for electrically controlled nanoparticle release. Langmuir 27:6316–6322
Sharma M, Waterhouse GIN, Loader SWC, Garg S, Svirskis D (2013) High surface area polypyrrole scaffolds for tunable drug delivery. Int J Pharm 443:163–168
Cho SJ, Kim HJ, Lee JH, Choi HW, Kim HG, Chung HM, Do JT (2010) Silica coated titania nanotubes for drug delivery system. Mater Lett 64:1664–1667
Yang N, Chen X, Ren T, Zhang P, Yang D (2015) Carbon nanotube based biosensors. Sensors Actuators B Chem 207(Part A):690–715
Barik MA, Dutta JC (2014) Fabrication and characterization of junctionless carbon nanotube field effect transistor for cholesterol detection. Appl Phys Lett 105:053509-1–053509–5
Wu Z, W-q S, Feng T, Tang SW, Li G, Jiang K-l, Xu S-y, Ong CK (2015) Imaging of soft material with carbon nanotube tip using near-field scanning microwave microscopy. Ultramicroscopy 148:75–80
Barghi SH, Tsotsis TT, Sahimi M (2014) Chemisorption, physisorption and hysteresis during hydrogen storage in carbon nanotubes. Int J Hydrog Energy 39(3):1390–1397
Hashmi SG, Moehl T, Halme J, Ma Y, Saukkonen T, Yella A, Giordano F, Decoppet JD, Zakeeruddin SM, Lund P, Gratzel M (2014) A durable SWCNT/PET polymer foil based metal free counter electrode for flexible dye-sensitized solar cells. J Mater Chem 2:19609–19615
Ren H, Pyo S, Lee J-I, Park T-J, Gittleson FS, Leung FCC, Kim J, Taylor AD, Lee H-S, Chae J (2015) A high power density miniaturized microbial fuel cell having carbon nanotube anodes. J Power Sources 273:823–830
Shvedova A, Castranova V, Kisin E, Schwegler-Berry D, Murray A, Gandelsman V, Maynard A, Baron P (2003) Exposure to carbon nanotube material: assessment of nanotube cytotoxicity using human keratinocyte cells. J Toxicol Environ Health 66:1909–1926
Manna SK, Sarkar S, Barr J, Wise K, Barrera EV, Jejelowo O, Rice-Ficht AC, Ramesh GT (2005) Single-walled carbon nanotube induces oxidative stress and activates nuclear transcription factor-κB in human keratinocytes. Nano Lett 5:1676–1684
Muller J, Huaux F, Moreau N, Misson P, Heilier J-F, Delos M, Arras M, Fonseca A, Nagy JB, Lison D (2005) Respiratory toxicity of multi-wall carbon nanotubes. Toxicol Appl Pharmacol 207:221–231
Cui D, Tian F, Ozkan CS, Wang M, Gao H (2005) Effect of single wall carbon nanotubes on human HEK293 cells. Toxicol Lett 155:73–85
Ivanova VT, Katrukha GS, Timofeeva AV, Ilyna MV, Kurochkina YE, Baratova LA, Sapurina IY, Ivanov VF (2011) The sorption of influenza viruses and antibiotics on carbon nanotubes and polyaniline nanocomposites. J Phys Conf 291:012004
Zwilling V, Darque-Ceretti E, Boutry-Forveille A, David D, Perrin MY, Aucouturier M (1999) Structure and physicochemistry of anodic oxide films on titanium and TA6V alloy. Surf Interface Anal 27:629–637
Gong D, Grimes CA, Varghese OK, Hu W, Singh RS, Chen Z, Dickey EC (2001) Titanium oxide nanotube arrays prepared by anodic oxidation. J Mater Res 16:3331–3334
Liu G, Wang K, Hoivik N, Jakobsen H (2012) Progress on free-standing and flow-through TiO2 nanotube membranes. Sol Energy Mater Sol Cells 98:24–38
Xiao X, Yang L, Guo M, Pan C, Cai Q, Yao S (2009) Biocompatibility and in vitro antineoplastic drug-loaded trial of titania nanotubes prepared by anodic oxidation of a pure titanium. Sci China B Chem Life Sci Earth Sci 52:2161–2165
Neupane MP, Park IS, Bae TS, Yi HK, Uo M, Watari F, Lee MH (2011) Titania nanotubes supported gelatin stabilized gold nanoparticles for medical implants. J Mater Chem 21:12078–12082
Popat KC, Eltgroth M, LaTempa TJ, Grimes CA, Desai TA (2007) Titania nanotubes: a novel platform for drug-eluting coatings for medical implants? Small 3:1878–1881
Wilmowsky CV, Bauer S, Lutz R, Meisel M, Neukam FW, Toyoshima T, Schmuki P, Nkenke E, Schlegel KA (2009) In vivo evaluation of anodic TiO2 nanotubes: an experimental study in the pig. J Biomed Mater Res B Appl Biomater 89:165–171
Baowan D, Sukchom W, Chayantrakom K, Satiracoo P (2011) Three possible encapsulation mechanics of TiO2 nanoparticles into single-walled carbon nanotubes. J Nanomater ID 857864:1–8
Babazadeh M, Gohari FR, Olad A (2012) Characterization and physical properties investigation of conducting polypyrrole/TiO2 nanocomposites prepared through a one-step “in situ” polymerization method. J Appl Polym Sci 123:1922–1927
Swami N, Cui Z, Nair LS (2011) Titania nanotubes: novel nanostructures for improved Osseointegration. J Heat Transf 133:034002
Noh K, Brammer KS, Kim SH, Choi C, Frandsen CJ, Jin S (2011) A new nano-platform for drug release via nanotubular aluminum oxide. J Biomater Nanobiotechnol 2:226–233
Kong Y, Ge H, Xiong J, Zuo S, Wei Y, Yao C, Deng L (2014) Palygorskite polypyrrole nanocomposite: a new platform for electrically tunable drug delivery. Appl Clay Sci 99:119–124
Gao W, Borgens RB (2015) Remote-controlled eradication of astrogliosis in spinal cord injury via electromagnetically-induced dexamethasone release from “smart” nanowires. J Control Release 211:22–27
Acknowledgments
The authors gratefully acknowledge the financial support provided by the Ministry of Higher Education and the Ministry of Health of Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is dedicated to Professor José H. Zagal on the occasion of his 65th birthday with appreciation of his studies on conductive polymer films.
Rights and permissions
About this article

Cite this article
Alshammary, B., Walsh, F.C., Herrasti, P. et al. Electrodeposited conductive polymers for controlled drug release: polypyrrole. J Solid State Electrochem 20, 839–859 (2016). https://doi.org/10.1007/s10008-015-2982-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-015-2982-9