
Abstract
Composite materials of Prussian Blue–polypyrrole (PB/PPy) were obtained via chemical redox process in mixed solution of iron (III), hexacyanoferrate (III), and pyrrole with chloride or nitrate supporting electrolyte. Synthesized composites in the form of a sedimented powder, or a film on the surface of Pt and ITO-coated glass, were characterized by various physical and electrochemical methods. Stability of PB/PPy films on Pt substrate was tested in electrocatalytic reaction of hydrogen peroxide reduction in weakly acidic medium (pH 6) as well as on ITO-coated glass substrate via electrochromic (spectroelectrochemical) measurements. Stability period of the amperometric response in 1 mM H2O2 solution for films on Pt substrate synthesized in chloride media is 20 times longer than that of pure PB films obtained electrochemically in potentiostatic mode. Morphology of PB/PPy films has been found to depend on the composition of supporting electrolyte. Synthesis of PB/PPy composites in nitrate electrolyte leads to formation of a high-quality morphology, whereas PB/PPy films obtained in chloride solutions are cracked. In conformity with this observation, the stability period of the H2O2 electroreduction for nitrate-synthesized films on Pt substrate was about 150 min, i.e., 80 times longer than that of pure Prussian Blue films without polymeric support and four times longer than that of chloride-synthesized films. Stability of the electrochromic response of PB/PPy films formed in nitrate media is 10 times higher than pure PB film.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Prussian Blue (PB), Fe4[Fe(CN)6]3, as a film on electrode surface represents an electroactive material due to its redox transformations to both partially (Prussian Green) or fully oxidized (Prussian Yellow) and reduced (Prussian White, PW) forms as a function of the electrode potential [1–3]. The reduced form of PB, PW, can act as electrocatalyst for reduction of hydrogen peroxide present in solution or formed as a product of enzyme-catalyzed reactions [4–10].
Electrodes modified by PB films can be easily obtained via different electrochemical procedures [1, 11–14], for example by potentiostatic reduction of equimolar mixture of iron (III) and ferricyanide salts [1, 12, 13]. However, such modified electrodes possess a crucial drawback for their sensor application: Their cathodic current in solutions of peroxide drops quickly and irreversibly, especially in neutral solutions [7, 9]. Even in dilute solution of hydrogen peroxide (e.g., 0.1 mM) PB, electrochemically modified electrodes diminish their catalytical activity by 5 % within about 8 min [15]. Stability of the electrochromic switch of PB films is greatly affected by the cation nature of the supporting electrolyte and cannot unambiguously be considered as high [2].
Electrodeposition of PB on the electrode surface coated with an organic layer was used with a hope to extend the stability period of such modified electrodes for electrochromic and sensor applications [16–18]. Another approach was to protect the PB film by various polymer layers [15, 19–24].
An alternative method to enhance the stability of PB-containing films is based on deposition of a PB/polymer composite via chemical (redox-reaction) procedure [15]. An equimolar mixture of Fe(III) and [Fe(CN)6]3− salts was used for oxidation of various monomers, including pyrrole. Such composite films demonstrated more extended periods of H2O2 electroreduction (in its 0.1 mM solution) compared to that for pure PB films.
These synthetic procedures have not been used extensively for studying the stability of the electrochromic switch of PB/polymer composites. Published results are not especially promising [25], the reason of this much lower stability of PB/polymer films remains unclear. These modified electrodes have only been characterized by means of electrochemical methods (redox response and peroxide electroreduction), while the structure and morphology of such PB/polymer composite films have not been studied.
Synthesis of modified electrodes in those studies was carried out in chloride-based supporting electrolyte, in line with the dominant tendency to use this electrolyte for pure PB films [1, 9, 12, 26, 27] and PB containing systems [23, 28, 29]. In this context, it is worth to note the conclusion by Itaya et al. [30] that the stability of the redox response of pure PB films in the background solution depends on the type of anion of the supporting electrolyte.
The actual work represents a study of the dependence of redox, and electrocatalytic and electrochromic properties of composite PB/polypyrrole (PB/PPy) films on various synthesis conditions, with thorough characterization of structural and morphological properties of these films. It was aimed at optimization of the synthesis procedure to reach a high-stability electrochemical (electrocatalytic and electrochromic) response of such modified electrodes.
Experimental
Synthesis of PB/PPy composites
Similar to the procedure in [15], PB/PPy composites were generated via a one-pot one-step redox reaction between the oxidizer (equimolar mixture of iron(III) and ferricyanide salts) and the reducing agent (pyrrole, Py, taken in excess) in their mixed aqueous solution, with addition of a background electrolyte. The acidity was sufficiently high to prevent the hydrolysis of the Fe(III) ion.
As in the previous study [15], chloride-based electrolyte, 0.1 M HCl + 0.1 M KCl, was tested first. Then, the same synthesis procedure was carried out in nitrate solution, 0.1 M HNO3 + 0.1 M KNO3.
FeCl3·6H2O (p.a., Carl Roth GmbH) salt was used in chloride media and Fe(NO3)3·9H2O (p.a., Carl Roth GmbH) in nitrate ones, combined with K3[Fe(CN)6] (p.a., Carl Roth GmbH) reagent in both cases. All solutions of reagents were freshly prepared. Pyrrole monomer (98 %, Alfa Aesar) was distilled under inert atmosphere (Ar, with vacuum line used for deoxygenation) every time prior to use. Initial solutions of reagents were mixed in the following order: first, iron(III) and ferricyanide salts, then Py solution was added (all starting solutions contained the same background electrolyte). The reaction was carried out at room temperature, 20 ± 1 °C.
As it was demonstrated in the synthesis of Pd/PPy composites via the same redox-reaction method [31, 32], it is advantageous to use low concentrations of reactants, resulting in an extended period of the composite formation. Therefore, in the actual study, the concentrations of the reaction mixture were 0.1 mM Fe3+, 0.1 mM [Fe(CN)6]3−, and 0.5 mM Py, compared to 2 mM Fe3+, 2 mM [Fe(CN)6]3−, and 1–20 mM Py in [15].
Electrodes and cells
Electrochemical measurements were performed in single-compartment three-electrode cell with coiled Pt wire as counter electrode and SCE (Jenway) as reference electrode. All potentials in the paper are given versus SCE. Cell was deoxygenated by means of vacuum line, and then it was kept under inert Ar atmosphere during all measurements. Electrochemical studies were performed with the use of PGSTAT30 (Autolab Eco Chimie), while spectroelectrochemical measurements were performed under potential control by PI-50 Pro (Elins).
PB/PPy composite and pure PB films were deposited at the surface of Pt and ITO-coated glass working electrodes.
Disk Pt electrodes (surface area: 0.01 cm2) soldered in glass were polished by diamante suspension (1 μm, Escil) on flap disk (FD 1, Escil), rinsed with ethanol and distilled water, and then electrode was ultrasound-treated in distilled water for 5 min.
Rectangular ITO-coated glass substrates (surface area about 1 cm2, resistance below 20 Ω−2, Präzisions Glas & Optik GmbH) were rinsed with ethanol, distilled water, and with concentrated solutions of NaOH and H2SO4. After that, ITO-coated glasses were ultrasound-treated in ethanol for 15 min and kept in drying box at 80 °C.
To modify the electrode surface with PB/PPy film, the electrode was placed in reaction mixture for 16–18 h. Then, it was rinsed thoroughly with distilled water.
Pure PB films were electrodeposited on the surface of Pt or ITO-coated glass substrate, for comparison of their redox activity and electrochemical stability with those of PB/PPy-modified electrodes. In this case, the synthesis procedure was performed electrochemically in potentiostatic mode (0.55 V vs. SCE for 100 s) in mixed aqueous solution: 10 mM Fe(NO3)3 + 10 mM K3[Fe(CN)6] with 0.1 M HNO3 + 0.1 M KNO3 as supporting electrolyte.
Methods of characterization of PB/PPy composites and PB films
Redox activity of synthesized PB/PPy composites immobilized on electrode surface was tested in the same chloride or nitrate background electrolyte which was used for their synthesis. Tests of the electrocatalytic activity of PB/PPy films in H2O2 solution were performed both with the use of the same background electrolyte and of phosphate buffer (pH 6.0).
Similar studies were carried out with PB films on electrode surface.
Redox activity of PB modified electrodes related to reversible transformation of PB to PW due to reduction of Fe(III) ions inside the PB lattice was characterized by cyclic voltammetry (CV) [1].
Stability tests of composite films were carried out either under conditions of the hydrogen peroxide electroreduction or in the course of multi-cycle spectroelectrochemical experiments. Electrochemical and spectroelectrochemical measurements were carried out at room temperature, 22 ± 1 °C.
Electroreduction of hydrogen peroxide at these modified electrodes was performed in potentiostatic conditions corresponding to the limiting-diffusion range (0 V).
Spectroelectrochemical measurements were realized in potentiodynamic mode (CV measurements) with synchronous measurements of absorbance at 720 nm which corresponds to the maximum of the PB absorption band. Special home-made three-electrode optical cell was used. Namely, working ITO-coated glass electrode with a composite or pure PB film deposited on its conducting surface was placed into 10-mm quartz cuvette in contact with working solution inside the cell. Both counter and tiny reference electrode contacted with the same solution via glass frits. Kinetic absorbance measurements (at 720 nm, registration time 1 s) were carried out by means of Lightwave II (Biochrom) spectrophotometer. Potential range in spectroelectrochemical CV measurements was between −0.145 V and 0.500 V vs. SCE, scan rate 100 mV/s. The film-coated electrode was subjected to such repeating CV treatment until the cathodic (or anodic) charge of the cycle has become lower than 25–30 % of the charge of the second cycle. This diminution of the redox charge of the film was found to correspond to the same degradation degree of the synchronically measured film absorbance. In this context, CV measurements could be used as a way to estimate the stability of the electrochromic transition of PB-containing films.
Morphology of PB/PPy films on the ITO-coated glass surface was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) by means of JEOL JSM-6400F and field-emission gun scanning electron microscope (FEG-SEM) JEOL JSM 6500F instruments at 15 or 20 kV accelerating voltage and Jeol JEM-2100 TEM with LaB6 source operating at accelerating voltage of 200 kV. Chemical analysis of samples was performed by means of Oxford Instruments EDS analyzer using INCA software. XRD spectra of such films were recorded at CPS 120 I.E. diffractometer with monochromatized Cu Kα radiation at 40 mA and 40 kV. Thickness of PB/PPy composite films obtained from TEM and SEM data and calculated via electrochemical and spectroscopic data was 100–150 nm.
Results and discussion
Synthesis of PB/PPy composites was performed as a one-step procedure inside mixed aqueous solution of 0.1 mM Fe(III), 0.1 mM [Fe(CN)6]3−, and 0.5 mM Py with the chloride or nitrate background electrolyte (pH 1). These ions form a complex Fe[Fe(CN)6] [33–35], its concentration in our conditions being of the order of 10−7 M.
Both Fe(III) and the complex Fe[Fe(CN)6] are able to oxidize pyrrole in mixed solution, with simultaneous generation of PB and PPy in the form of the PB/PPy composite, 1.5/n [Py]n·Fe4[Fe(CN)6]3:
Blue colored colloid solution is formed in a few hours. UV-visible spectra of reaction mixture (Fig. 1) demonstrate а new (arising with time) absorption band around 700 nm corresponding to PB [1, 2], as one can see from its comparison with the spectrum of a pure PB film on ITO-coated glass (inset in Fig. 1, curve 5). Sedimentation rate of the PB/PPy colloid depends on temperature and concentrations of reagents. Under our reaction conditions, a blue film on walls of the vessel and some amount of dark-blue precipitate in the powder form were formed in 14–16 h.

UV-visible spectra. Individual reagents in 10-mm quartz cuvette: 0.1 mM FeCl3 (1) and 0.1 mM K3[Fe(CN)6] (2) in 0.1 M HCl + 0.1 M KCl. PB/PPy reaction mixture (0.1 mM Fe3+, 0.1 mM [Fe(CN)6]3− and 0.5 mM Py in chloride medium) 1.5 h since the beginning of reaction (3). PB/PPy composite film on ITO-coated glass plate in 14 h since the beginning of reaction (4). Pure PB film electrodeposited on ITO-coated plate electrode (5)
Characterization of PB/PPy composite films
To modify an electrode surface with PB/PPy film, it was immersed into the reaction mixture for the whole reaction period. The PB/PPy film on the surface of electrode has a blue color. UV-visible spectra of such films on ITO-coated glass substrates demonstrate the PB absorption band around 700 nm (Fig. 1, curve 4). A lower intensity of the PB absorbance inside of the composite film (Fig. 1, curve 4) compared to that of the pure PB film formed by the electrodeposition procedure (Fig. 1, curve 5) may be attributed to the difference in their thicknesses as well as a lower content of PB inside the composite.
For characterization of the film structure deposited on the electrode surface, a special procedure was used to prepare samples for TEM analysis. After the end of the deposition process, the film on the walls of the vessel was thoroughly rinsed with distilled water. Then, the vessel was filled with ethanol (99 %) and treated ultrasonically for 10 to 15 min. It leads to formation of a homogeneous blue colloid solution. Drops of this solution were placed on TEM substrate, with subsequent solvent evaporation.
According to TEM images, the PB/PPy composite synthesized in chloride medium is composed of semitransparent polypyrrole globules of a uniform size (about 50 nm in diameter) with denser particles inside them (Fig. 2a). More detailed HR-TEM image (Fig. 2b) shows that this inorganic component consists of square elements of 50 nm in dimension, i.e., occupying most of the internal space of the globule, with a relatively thin polymer layer on the particle surface. Selected area electron diffraction (SAED) reveals that these inorganic particles represent PB crystals (Fig. 3а).

TEM (а) and HR-TEM (b) images of PB/PPy composites synthesized from 0.1 mM Fe3+, 0.1 mM [Fe(CN)6]3−, and 0.5 mM Py reaction mixture in chloride medium

SAED (a, b) of PB/PPy composite synthesized in chloride (а) and nitrate (b) media and analyzed on the TEM substrate. XRD spectra (c) of PB/PPy composite films on ITO-coated glass surface synthesized in chloride (1) and nitrate (2) media, in comparison with peaks of pure PB crystals (bars, JCPDF 052-1907). Other peaks in XRD spectra of the composites (1 and 2) correspond to ITO-coated glass substrate
Peaks in the XRD spectrum of the composite film deposited on ITO-coated glass surface from chloride medium (Fig. 3c) also correspond to the PB structure. The size of crystal elements inside this film estimated from the width of the <200> peak with the use of the Scherrer equation was about 30–35 nm.
Composite material generated by the same redox reaction inside nitrate electrolyte is deposited mainly on the walls of vessel as a compact film with a much stronger adhesion to their surface, while the amount of sedimented precipitate on the bottom is very small. TEM images (Fig. 4a) demonstrate that PB/PPy composite extracted from such film is composed mostly of rectangular PB elements of a bit greater dimension (about 100 nm) than those in chloride medium. An interesting feature is their semitransparence for TEM electrons (Fig. 4a). Similar to the composite obtained in chloride solutions, they are covered by a thin polypyrrole layer (Fig. 4).

TEM (a) HR-TEM (b) images of PB/PPy composites synthesized from 0.1 mM Fe3+, 0.1 mM [Fe(CN)6]3−, and 0.5-mM Py reaction mixture in nitrate medium
It may be observed in HR-TEM images (Figs. 2b and 4b) that individual atomic rows inside PB single crystals are parallel (or perpendicular) both to each other and to external boundaries of the crystal. Moreover, some atomic rows in Fig. 4b may be traced within the whole dimension of the image, i.e., at the distance over 50 nm. Such high structural order as well as a square shape with straight-lined boundaries of all inorganic elements (Fig. 4a) led us to conclude of their single-crystal type.
Attribution of these denser elements to PB crystals was confirmed by SAED (Fig. 3b) and XRD (Fig. 3c, curve 2) techniques. The size of PB crystalline fragments estimated from the XRD peak at 17° (<200>) was about 28 nm, i.e., comparable to that for films deposited in chloride medium.
The peak positions in XRD spectra corresponding to PB crystals inside PB/PPy films on Pt substrate are identical to those on ITO-coated glass substrate. Thus, one can conclude that structural properties of PB crystals inside these composite layers are independent of the solid substrate and are similar for films deposited from chloride or nitrate medium.
SEM images (Fig. 5) reveal a surprising difference in the morphology of PB/PPy films on ITO-coated glass surface depending on the electrolyte. It turned out that deposition from chloride electrolyte (which is dominantly used for synthesis of PB containing materials) results in films with numerous cracks (Fig. 5a). On the contrary, films of the PB/PPy composite obtained in nitrate electrolyte demonstrate quite a different morphology: a very uniform and compact layer (Fig. 5b).

SEM images of PB/PPy composite films on ITO-coated glass support obtained in chloride (a) and nitrate (b) electrolytes
One should mention that nitrate-containing electrolyte has been used for depositing pure PB nanoparticles on the surface of carbon-ceramic electrodes [36].
EDX analysis shows that iron is present in samples obtained both in chloride and nitrate media, the ratio of iron to nitrogen being about the same (1:10–12), i.e., they represent chemically similar composite materials.
Electrochemical properties of PB/PPy composites
Electrochemical activity of Pt electrode covered by PB/PPy film was tested initially in the corresponding background electrolyte and then in phosphate buffer. As it shown in Fig. 6, there are redox peaks in CV curves related to the PB/PW transition (Fe4[Fe(CN)6]3/K4Fe4[Fe(CN)6]3) [1]. The sharp form of these peaks and low peak separation points to a regular structure of PB crystals. A higher capacitive current around the PB peaks is due to electroactivity of polypyrrole component of the film.

Stability of PB-containing films has been tested with respect to hydrogen peroxide electroreduction. Similar study had been carried out earlier in mild conditions of the 0.1 mM peroxide concentration [15]. Composite films prepared via the actual synthesis procedure turned out to be too stable under these conditions to record their degradation period since it was too long. Therefore, this parameter was determined by tests for 10 times higher peroxide concentration, 1 mM, in the same buffer solution. Even under such harsh conditions, our films retained their electrocatalytic activity for a relatively extended time.
Namely, films on Pt substrate obtained in chloride solution hold their redox response on a constant level for about 40 min (Fig. 7, curve 2), i.e., 5 times greater compared to pure PB film in 0.1 mM H2O2 [15] and 20 times greater compared to pure PB film in 1 mM H2O2 (Fig. 7, curve 1).

Current density of H2O2 electroreduction in 1 mM hydrogen peroxide with phosphate buffer, pH 6, on Pt electrode modified with pure PB (1) or PB/PPy composite film synthesized in chloride electrolyte (2) or in nitrate electrolyte (3). Current densities for PB/PPy film in 1 mM hydrogen peroxide with phosphate buffer, pH 6 (4), and in the same buffer without H2O2 (5) within the initial time interval (insert)
Under the same severe conditions of 1 mM peroxide with pH 6 buffer, the stability period of PB/PPy composite films synthesized in nitrate electrolyte turned out to be even longer, 150 min (Fig. 7, curve 3), i.e., 80 times greater than that of pure PB sensor and four times greater than that of composite films obtained in chloride medium.
During a more extended period, the current of the peroxide electroreduction is diminishing progressively. In parallel, the voltammetric peak currents of PB/PW transformation are becoming lower, while UV-visible data show a decrease of the PB content inside the film. This loss of PB in the film correlates well with decrease of the iron content inside the film in EDX data. Nevertheless, diffraction pattern of PB is still observed in XRD spectra. It means that PB retains its crystalline structure even after decrease of the peroxide reduction current.
SEM images show that the difference in morphology of PB/PPy films synthesized in chloride and nitrate electrolytes plays an important role in stability of the film on the surface of electrode. In the case of PB/PPy (Cl−) films a significant fraction of the film peeled off from the electrode surface due to penetration of hydrogen peroxide solution through cracks (formed already in the course of the film deposition stage) during electroreduction procedure (Fig. 8a), whereas one can see only minor damages in the structure of PB/PPy (NO3 −) composite films after electroreduction of peroxide (Fig. 8b). One may conclude on the basis of comparison of SEM and XRD data for films before and after the electrocatalytic activity that the principal cause of the decrease of the peroxide electroreduction current is a progressive decomposition of the film.

SEM images of PB/PPy films on ITO-coated glass surface after electroreduction in 1-mM solution of H2O2. Composite films were obtained in chloride (а) or nitrate (b) medium
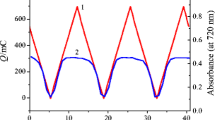
Stability of the electrochromic response of PB/PPy films synthesized in nitrate media on ITO-coated glass substrate was tested by means of the spectroelectrochemical procedure (in contact with background nitrate solution). Redox transformation PB/PW inside these films is accompanied by color transformation from blue to practically colorless (Fig. 9). Multi-cycle repetition of this spectroelectrochemical procedure leads to progressive diminution of both the redox charge in CV curves and the absorbance at 720-nm wavelength. Stability degree of such transformation was characterized by the diminution of the redox charge (cathodic or anodic) of the composite film transformation during multi-cycle CV potential variation (scan rate 100 mV/s) in the range between −0.145 and +0.500 V. Figure 10 demonstrates the variation of the stability degree of PB/PPy films prepared via redox procedure in nitrate medium, in comparison with that for the pure PB film. Stability degree of the composite film during 3000 cycles of CV measurements decreased only by 30 % (curve 1), whereas the stability degree of the pure PB film was rapidly diminishing already within the first 300 cycles (curve 2). Electrochromic transformation of these films (estimated from the variation of the absorbance spectrum) showed a similar stability. Degradation of the electrochromic signal occurs due to an instability of the film in the PW state. After such spectroelectrochemical treatment, PB-containing films look spotted since some pieces of the film were peeled off from the electrode surface.

Spectroelectrochemical measurements of PB/PW transformation in PB/PPy film synthesized in nitrate medium on ITO-coated glass electrode. CV response of the film (100 mV/s) (1) and absorbance changes (at 720 nm) in the course of this CV potential variation (2). Photos demonstrate the colors of the composite film in its reduced (−0.145 V) and oxidized (+0.500 V) states

Stability degree variation of the composite PB/PPy film (1) and the pure PB film (2) on the ITO-coated glass electrode
Conclusions
In this study, we have elaborated the one-step method of the chemical deposition of PB-polypyrrole films on a conducting or insulating solid substrate (including an electrode surface) from the mixed solution of pyrrole, iron(III), and ferricyanide salts proposed in [15]. These materials have been characterized by a set of physical and electrochemical methods. It has been found that such films consist of composite particles where small PB single crystals (semitransparent for TEM electrons) are incorporated into PPy surrounding.
Drastic dependence of the morphology of these films on the anion of the “background” electrolyte has been discovered. Realization of the synthesis procedure in chloride electrolyte leads to formation of cracked films, whereas the use of nitrate solution results in high-quality films without cracks.
It is important to mention that PB inside the composite film retains its crystalline structure even after its long-time functioning as electrocatalyst in 1-mM hydrogen peroxide solution, the size of PB crystals remaining unaffected (according to XRD spectra). It means that the progressive loss of its catalytic properties is not due to a progressive dissolution of the reduced form, PW. Our observations testify in favor of the explanation that the presence of cracks of the surface of the film can be the principal reason of this electroactivity loss, due to partial peeling of the film from the electrode surface during this process.
These promising results have been obtained for PB/PPy composite films deposited on Pt substrate. For ITO-coated glass substrates, we have observed a high stability of the electrochromic transformation of PB/PPy films formed in nitrate electrolyte, compared to pure PB films. Stability period of PB/PPy films is 10 times greater than that for pure PB films.
References
Itaya K, Akahoshi H, Toshima S (1982) Electrochemistry of Prussian Blue modified electrodes: an electrochemical preparation method. J Electrochem Soc 129(7):1498–1500
Itaya K, Ataka T, Toshima S (1982) Spectroelectrochemistry and electrochemical preparation method of Prussian Blue modified electrodes. J Am Chem Soc 104:4767–4772
Itaya K, Uchidа I, Neff V (1986) Electrochemistry of polynuclear transition metal cyanides: Prussian Blue and its analogues. Acc Chem Res 19:162–168
Boyer A, Kalcher K, Pietsch R (1990) Voltammetric behavior of perborate on prussian-blue-modified carbon paste electrodes. Electroanalysis 2(2):155–161
Derwinska K, Miecznikowski K, Koncki R, Kulesza PJ, Glab S, Malik MA (2003) Application of Prussian Blue based composite film with functionalized organic polymer to construction of enzymatic glucose biosensor. Electroanalysis 15(23-24):1843–1849
Zhang J, Li J, Yang F, Zhang B, Yang X (2009) Preparation of Prussian blue@Pt nanoparticles/carbon nanotubes composite material for efficient determination of H2O2. Sens Actuators B 143(1):373–380
Karyakin AA (2001) Prussian Blue and its analogues: electrochemistry and analytical applications. Electroanalysis 13(10):813–819
Ricci F, Amine A, Palleschi G, Moscone D (2003) Prussian Blue based screen printed biosensors with improved characteristics of long-term lifetime and pH stability. Biosens Bioelectron 18(2–3):165–174
De Mattos IL, Gorton L, Ruzgas T (2003) Sensor and biosensor based on Prussian Blue modified gold and platinum screen printed electrodes. Biosens Bioelectron 18(2–3):193–200
Ricci F, Palleschi G (2005) Sensor and biosensor preparation, optimisation and applications of Prussian Blue modified electrodes. Biosens Bioelectron 21:389–407
Lisowska-Oleksiak A, Nowak AP, Jasulaitiene V (2006) Poly(3,4-ethylenedioxythiophene)-Prussian Blue hybrid material: evidence of direct chemical interaction between PB and pEDOT. Electrochem Commun 8(1):107–112
Mortimer RJ, Rosseinsky DR (1983) Electrochemical polychromicity in iron hexacyanoferrate films, and a new film form of ferric ferricyanide. J Electroanal Chem 151(1–2):133–147
Mortimer RJ, Rosseinsky DR (1984) Iron hexacyanoferrate films: spectroelectrochemical distinction and electrodeposition sequence of ‘soluble’ (K+-containing) and ‘insoluble’ (K+-free) Prussian Blue, and composition changes in polyelectrochromic switching. J Chem Soc Dalton Trans 2059-2061
Lisowska-Oleksiak A, Nowak AP (2007) Metal hexacyanoferrate network synthesized inside polymer matrix for electrochemical capacitors. J Power Sources 173(2):829–836
Borisova AV, Karyakina EE, Cosnier S, Karyakin AA (2009) Current-free deposition of Prussian Blue with organic polymers: towards improved stability and mass production of the advanced hydrogen peroxide transducer. Electroanalysis 21(3-5):409–414
Somani P, Radhakrishnan S (1998) Electrochromic response in polypyrrole sensitized by Prussian blue. Chem Phys Lett 292:218–222
Lu W, Wallace GG, Karayakin AA (1998) Use of Prussian Blue/conducting polymer modified electrodes for the detection of cytochrome C. Electroanalysis 10(7):472–476
Zhao H, Yuan Y, Adeloju S, Wallace GG (2002) Study on the formation of the Prussian blue films on the polypyrrole surface as a potential mediator system for biosensing applications. Anal Chim Acta 472:113–121
García-Jareño JJ, Navarro-Laboulais J, Vicente F (1996) Charge transport in Prussian blue films deposited on ito electrodes. Electrochim Acta 41(6):835–841
Guo Y, Guadalupe AR, Resto O, Fonseca LF, Weisz SZ (1998) Chemically derived Prussian blue sol − gel composite thin films. Chem Mater 11(1):135–140
Koncki R, Wolfbeis OS (1998) Composite films of Prussian Blue and N-substituted polypyrroles: fabrication and application to optical determination of pH. Anal Chem 70:2544–2550
Curulli A, Valentini F, Orlanduci S, Terranova ML, Palleschi G (2004) Pt based enzyme electrode probes assembled with Prussian Blue and conducting polymer nanostructures. Biosens Bioelectron 20(6):1223–1232
Vidal JC, Espuelas J, Garcia-Ruiz E, Castillo JR (2004) Amperometric cholesterol biosensors based on the electropolymerization of pyrrole and the electrocatalytic effect of Prussian-Blue layers helped with self-assembled monolayers. Talanta 64:655–664
Garjonyte R, Malinauskas A (2000) Amperometric glucose biosensors based on Prussian Blue– and polyaniline–glucose oxidase modified electrodes. Biosens Bioelectron 15(9–10):445–451
Wang H, Guo C, Zhou S, Hu X, Hu Y, Li F, Miao Y (2012) One-step synthesis and self-organization of polypyrrole ultrathin films inlayed with Prussian blue nanoparticles induced by a drop of toluene solution on water surface. Thin Solid Films 520:2026–2031
Neff VD (1978) Electrochemical oxidation and reduction of thin films of Prussian Blue. J Electrochem Soc 125(6):886–887
Mortimer RJ, Reynolds JR (2005) In situ colorimetric and composite coloration efficiency measurements for electrochromic Prussian blue. J Mater Chem 15:2226–2233
Lupu S, Mihailciuc C, Pigani L, Seeber R, Totir N, Zanardi C (2002) Electrochemical preparation and characterisation of bilayer films composed by Prussian Blue and conducting polymer. Electrochem Commun 4(10):753–758
Malik MA, Kulesza PJ, Wlodarczyk R, Wittstock G, Szargan R, Bala H, Galus Z (2005) Formation of ultra-thin prussian blue layer on carbon steel that promotes adherence of hybrid polypyrrole based protective coating. J Solid State Electrochem 9(5):403–411
Itaya K, Uchidа I (1986) Nature of Intervalence charge-transfer bands in Prussian Blues. Inorg Chem 25:389–392
Vasilyeva SV, Vorotyntsev MA, Bezverkhyy I, Lesniewska E, Heintz O, Chassagnon R (2008) Synthesis and characterization of palladium nanoparticle/polypyrrole composites. J Phys Chem C 112(50):19878–19885
Zinovyeva VA, Vorotyntsev MA, Bezverkhyy I, Chaumont D, Hierso JC (2011) Highly dispersed palladium–polypyrrole nanocomposites: in-water synthesis and application for catalytic arylation of heteroaromatics by direct C–H bond activation. Adv Funct Mater 21(6):1064–1075
Ibers JA, Davidson N (1951) On the interaction between hexacyanatoferrate(III) ions and (a) hexacyanatoferrate(II) or (b) iron(III) ions. J Am Chem Soc 73:476–478
Singleton DL, Swinehart JH (1967) A kinetic study of the hexaaquoiron( III)-hexacyanoferrate(III) complex. Inorg Chem 6(8):1539–1536
Walker RG, Watkins KO (1968) A study of the kinetics of complex formation between hexacyanoferrate(III) ions and lron(lIl) to form FeFe(CN)6 (Prussian Brown). Inorg Chem 7(5):885–888
Razmi H, Mohammad-Rezaei R, Heidari H (2009) Self-assembled Prussian Blue nanoparticles based electrochemical sensor for high sensitive determination of H2O2 in acidic media. Electroanalysis 21(21):2355–2362
Acknowledgments
The study was carried out with support of the Russian Ministry of Education and Science (project 2014-14-576-0056-058, agreement 14.574.21.0004).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Talagaeva, N.V., Zolotukhina, E.V., Bezverkhyy, I. et al. Stability of Prussian Blue–polypyrrole (PB/PPy) composite films synthesized via one-step redox-reaction procedure. J Solid State Electrochem 19, 2701–2709 (2015). https://doi.org/10.1007/s10008-015-2951-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-015-2951-3