
Abstract
The cathodic processes of cobalt ion in ionic liquids consisting in binary mixtures of choline chloride (ChCl) with urea, ethylene glycol, malonic acid or oxalic acid were investigated using cyclic voltammetry and electrochemical impedance spectroscopy. The as-received hexahydrate cobalt chloride has been the precursor of cobalt ionic species. It was found that the reduction mechanism of Co2+ in ChCl-urea and ChCl-ethylene glycol is a quasi-reversible (or with high degree of irreversibility at high scan rate) and diffusion controlled process, whereas in ChCl-carboxilic acid solvents the cathodic process of Co2+ is overlapped with reduction of protons. By using Pt and vitreous carbon electrodes, one has computed the value for the Co2+ diffusion coefficient in ChCl-urea at 80 °C; in comparison with values obtained in aqueous solutions, this is up to two orders of magnitude lower. The paper also describes preliminary experiments concerning electrodeposition of cobalt thin films on copper substrate from the four ionic liquid analogue solvents containing choline chloride. The crystalline morphology and structural constitution of Co deposits were examined by scanning electron microscopy, energy dispersive X-ray spectroscopy, and X-ray diffraction. Good results regarding the quality and cobalt content in films were obtained for Co deposits using ChCl-urea or ChCl-ethylene glycol mixtures as solvents. The behaviour of Co films grown from these media was typical for a ferromagnetic material.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The electrodeposition of Co and its alloys attracted a great interest in view of applications related to the production of corrosion-resistant alloys, sensors, heterogeneous catalysts or the use in energy storage. Thin and ultra-thin Co layers have also an important role in magneto-optic recording media applications, including the high density data storage parts integrated in functional micro or nanodevices (MEMS/NEMS). The electrodeposition of Co and its alloys has been carried out normally from aqueous solutions, and a number of electrolytes have been used, mostly based on chloride baths [1–4]. Cobalt sulphate was also used as precursor in baths for Co and Co alloys electrodeposition [5–7]. Many research programmes have been developed in order to substitute cobalt electrodeposition from aqueous solutions (to eliminate the intense hydrogen evolution) but the results were not always satisfactory as far as the current efficiency is concern. The use of water-free solvents with a wide electrochemical window seems to be the normal way to progress. Some promising results were reported by Cui et al. [8] who investigated a DMF-based bath for obtaining Co electrodeposit, while Suzaki and Watanabe [9] reported a comparative study of Co films prepared from five kinds of organic solvents (ethanol, formamide, N-methyl formamide, N,N-dimethyl formamide and dimethyl sulfoxide). Yamamoto et al. [10] suggested that an electrolytic medium with ethylene glycol may also be worth trying.
Ionic liquids are promising electrolytes for electrodeposition of various metals, since they have some common properties similar to organic electrolytes and to high-temperature molten salts, such as extended working temperature range, wide electrochemical window and good chemical stability. They also have the advantage that act as solvent and as electrolyte at the same time. The main drawback is the high cost involved, although the electrodeposition process is still cheaper than other high quality film deposition methods, such as sputtering and chemical vapour deposition. In the last years, the successful electrodeposition of metallic cobalt from CoCl2 precursor dissolved in ionic liquids containing 1-ethyl-3-methylimidazolium chloride (EMIC) has been reported; acidic mixtures based on chloroaluminate (AlCl3-EMIC) [11] or chlorozincate (ZnCl2-EMIC) [12, 13] were used as solvents. Chen et al. [12] observed that cobalt deposition occurs via three-dimensional instantaneous nucleation with diffusion-controlled growth of the nuclei. The 1-butyl-methylimidazolium salts, such as AlCl3-[BMIM]Cl and AlCl3-[BMIM]PF6 containing ionic liquids, were studied for Co films deposition by Freyland [14]. Various baths containing methylimidazolium salts were also investigated [15–18]. Schaltin et al. [16] used cyclic voltammetry and absorption spectrophotometry to examine the complex formation of cobalt (II) in the ionic liquids based on [BMIM]Cl and [BMIM][TFSI]. These authors have observed a round nodular morphology of micrometre size for the formed Co coating. Ali et al. [19] established that a low concentration of electroactive species CoCl3 − and a high concentration of inactive species CoCl4 2− (tetrahedral chloro complex) exist at 110 °C in CoCl2:N-butyl-pyridinium chloride mixture if the molar ratio exceeds 1:1.8 molar ratio.
Katayama, Fukui and Miura [20–22] showed that the electrodeposition of cobalt can be performed in 1-n-butyl-1-methylpyrrolidinum bis-trifluoromethyl sulfonyl imide ([BMP]TFSI). The large overpotential for the electrodeposition (1.5 V) was considered related to the structure of electric double layer; in particular, the layer formed by adsorption of BMP+ cation on the electrode surface in the negative potential region may inhibit not only the charge transfer but also some processes as nucleation, and surface diffusion. Ispas et al. [23] showed that Co and Co alloys may be deposited from [BMP]TFSA. Nevertheless, from the point of view of practical application, the traditional ionic liquids with imidazolium or pyrrolidinium derivates have high costs and have also potentially toxicological and purity issues.
In the last decade, the room temperature ionic liquids containing choline chloride (IUPAC name: (2-hydroxy-ethyl)-trimethyl ammonium chloride) were used as electrolyte to investigate the electrodeposition of metallic cobalt. In general, this new generation of ionic liquids, called also ‘deep eutectic solvents’ (DESs) supposes the use of mixtures of quaternary ammonium salts (mostly choline chloride, ChCl) with amides, glycols or carboxylic acids as hydrogen bond donors [24]. In this regard, the study in a recent paper [25] about the electrochemical properties of ChCl-urea can be mentioned. Eutectic compositions with a definite molar ratio of components (1:2 or 1:1) are easy to be prepared as solvent for deposition baths. Owing to the environmentally friendly properties, they have a low environmental impact ought to their biodegradability. However, their viscosity is generally higher than that of traditional ionic liquids, thus requiring relative high working temperature to improve the mass transport and electrodeposition kinetics.
Up to this moment, one can find that only few studies on electrodeposition of Co in DES ionic liquids based on choline chloride have been conducted. Films of Co, Sm and Sm-Co were electrodeposited on vitreous carbon [26] and copper [27] from the ChCl-urea (1:2 molar ratio) with CoCl2 content, having applications as homogeneous hard magnetic deposits of controlled composition. Using the same electrolyte and glass/Ti/Au or glass/ITO electrodes, Guillamat et al. [28] have investigated Co-Pt alloys coatings, as films for permanent magnets. The authors in references [26–28] have been able to detect the potential domain at which cobalt begins to electrodeposit. The temperature was selected at 70 °C to favour low viscosity and high electrical conductivity of the ionic liquid solvent. Recently, cyclic voltammetry at 100 °C has been used to study the reduction mechanism on tungsten electrode in ChCl-urea (1:2) + CoCl2 electrolyte [29]. The effects of electrode potential and temperature on deposit morphology and current efficiency of cobalt deposit were investigated. It is worth to notice that the cobalt chloride hexahydrate salt used as metal precursor in [26–29] was maintained in a stove prior to dissolution in ChCl-urea, this being considered a standard procedure that assures a maximum dehydration of the precursor.
However, as Abbott et al. showed [30], the limited range of concentration for anhydrous metal salt which forms ionic liquid with choline chloride restricts the use of such media for a variety of important electrochemical technologies. Binary mixtures of choline chloride with hydrated metal salts, including hydrated cobalt chloride, have been shown to form eutectic liquids that are air and moisture stable [31]. Srivastava et al. [32] electrodeposited cobalt on brass substrate at 70 °C using a mixture comprising choline chloride and cobalt chloride in the molar ratio of 1:2. These authors observed fine equiaxed crystallite morphology of Co coating, whereas acicular flakes were resulted in Co film prepared from aqueous electrolyte. Because binary mixtures of choline chloride with metal halides are not exactly ionic liquids, they are considered ionic liquid analogues [30]. Similarly, we can consider electrolytes formed by dissolution of hexahydrate cobalt dichloride in ChCl-based binary deep eutectic solvents (mixtures with urea, ethylene glycol, malonic acid or oxalic acid) as ionic liquid analogues.
You et al. [33] showed that Ni-Co alloy films can be obtained at room temperature on brass substrate from ChCl-ethylene glycol eutectic by adding nickel chloride and cobalt chloride as hexahydrate salts. Using these systems as baths, it seems that the presence of water molecules from hydrated salts is beneficial and the operational cost of electrodeposition is reduced, thus stimulating technological applications.
The aim of this paper is to investigate the electrochemical behaviour of cobalt ion, introduced as hexahydrate cobalt chloride, in four ChCl-containing binary mixtures: ChCl-urea (1:2), ChCl-ethylene glycol (1:2), ChCl-malonic acid (1:1) and ChCl-oxalic acid (1:1). We focused on these air and moisture stable ionic liquid analogues containing hydrated cobalt salt, as in our opinion they have great potential to be applied in electrodeposition technology in the future. We report preliminary results regarding cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements with Pt or glassy carbon electrode, as well as preparation and characterization (morphology, microstructure, magnetic properties) of electrodeposited Co thin films on copper substrate. To avoid the influence of the additives on the morphology and structure, the additive-free electrolytes were tested.
Experimental part
Choline chloride (98 wt% ChCl), cobalt (II) chloride hexahydrate, urea, ethylene glycol (EG), malonic acid (MA) and dihydrate oxalic acid (OxA), all from Aldrich, were used as received. In order to prepare the four types of ionic liquid eutectics as solvents, the mixtures of reagents in 1:2 molar ratio (of ChCl-urea and ChCl-EG) or in 1:1 molar ratio (of ChCl-MA and ChCl-OxA) were heated and were stirred at 90 °C for 30 min. The electrolytes with various concentrations of CoCl2 · 6H2O (from 0.01 to 1 M) were obtained by dissolving the appropriate amounts of cobalt precursor in already prepared ionic liquid. Values of densities determined previously [34] were used to compute the molarities at every working temperature. To ensure the dissolution of the salt and homogenization, the mixtures were heated again at 90 °C with permanent stirring. Their colour turned blue, which is specific for the presence of Co(II) species; the gradual transition from light blue (for diluted electrolyte) to dark blue colour is due to increase in CoCl2 · 6H2O content.
A Zahner Electrik IM6e potentiostat/galvanostat provided with a frequency analyzer was used for cyclic voltammetry (3–200 mV s−1 scan rates) and EIS measurements (10 mV sine waveform perturbation to the electrode potential, while scanning the modulus of impedance and the phase shifted in the frequency range 100–50 mHz). A plate (Pt, 0.5 cm2) or a disk (Pt, 0.07 cm2; glassy carbon, 0.07 cm2) was used as working electrode in the electrochemical cell. Pt plate as auxiliary electrode and Ag wire as quasi-reference electrode were used, respectively. All the potential values in the text are reported to this electrode. Before every electrochemical measurement, the working electrode was subjected to proper chemical pre-treatments for cleaning and removing the oxide or impurities: immersion in nitric acid solution (1:1) for a few minutes, polishing with 0.15 μm alumina suspension, rinsed with running water and Milli-Q water and dried.
Experiments involving electrolyses for Co electrodeposition on copper foil substrates (8–12 cm2 surface area) were carried out in non-stirred baths with controlled electrode potential or current. After deposition, the specimens were removed from the electrolyte, rinsed with water and acetone and air-dried. The film thickness was determined by weighing the mass gain after deposition. Film morphology and crystalline structure were investigated by scanning electron microscopy (Carl Zeiss EVO50) and X-ray diffractometry (Bruker AXS D8 ADVANCE employing Cu anode and kαNi filter), respectively. Energy-dispersive X-ray (EDX) device with Mn K resolution of 133 eV was used for elemental microanalysis. The peaks observed in the diffractograms were indexed by comparison with JCPDS database cards (no. 05–0727 for Co film and no. 04–0836 for Cu substrate). The hysteresis loops acquired during the magnetic measurements of the cobalt films were determined by a vibrating sample magnetometer (VSM-Lake Shore); the samples were placed both parallel and perpendicular to the magnetic field.
Results and discussion
Voltammetric behaviour of Co2+ ion in ionic liquids containing choline chloride
Electrode processes showing the electrochemical behaviour of cobalt ion were analyzed by cyclic voltammetry technique in four kinds of binary mixture of choline chloride with hydrogen bond donor compound: urea, ethylene glycol (EG), malonic acid (MA) and oxalic acid (OxA). Because in such studies it is important to know the electrochemical window of neat DES ionic liquid, CV curves of the basic media without metal precursor were previously plotted [35]. The corresponding electrochemical window in such curves becomes larger if the scan rate is increased (in the range 5–200 mV s−1) and this influence was monotonous. Using platinum electrodes, we obtained maximum values of electrochemical window up to 2.2, 2.1, 2 and 1.5 V for ChCl-urea, ChCl-EG, ChCl-MA or ChCl-OxA, respectively. The corresponding maximum values using glassy carbon (GC) electrode in the same media are greater than for platinum, reaching 3.1, 2.8, 2.2 and 2 V, respectively. All electrochemical window values are much larger than the corresponding value for an aqueous electrolyte, offering an increased capability for the investigated processes and allowing the obtaining of cobalt deposits with higher efficiency.
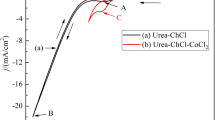
Cyclic voltammetry measurements were carried out in ChCl-urea eutectic with various concentrations of CoCl2 · 6H2O (up to 1 M Co2+). A selection of the obtained CV curves with first potential scan in a negative direction is presented in Fig. 1a–d.

CVs in ChCl-urea with 0.1 M CoCl2 (a, b) and 0.25 M CoCl2 (c, d) at 80 °C, for Pt (a, c) and GC (b, d) electrodes
Figure 1a shows a cathodic plateau of current (or an ill-defined peak) on Pt between −0.7 and −1.1 V vs. Ag, while in Fig. 1b–d, more pronounced cathodic peaks occur either for Pt or GC electrodes. In general, the shape of CV curves recorded using both kinds of electrodes is similar, with the start of cathodic current increasing at around −0.7 V vs. Ag. At a more negative potential, the deposition of Co is followed by a continuous increase of current, assigned to joint reduction of cobalt and choline ionic species. On the anodic branch of CVs depicted in Fig. 1, the well-defined peak recorded between −0.1 and +0.4 V vs. Ag is certainly related to the anodic dissolution of Co film. A supplementary oxidation process within potential range from +0.7 to +1.1 V vs. Ag is also observed as a wave or a distinct peak; we believe that it may be due to subsequent oxidation of Co2+ to Co3+, but a characterization of this process was not the object of this preliminary study. The final increase of anodic current is attributed to the oxidation of Cl− ions from solvent. Increasing the concentration up to 1 M Co2+, the processes are better evidenced on both types of electrodes.
The characteristics of reduction/oxidation pair for Co2+ ion are in a good agreement with previous reported voltammetric results in ChCl-urea DES ionic liquid [26, 27, 29]. We agreed with the assumption from literature that Co2+ ion exists as chloro complex ionic species, CoClx 2-x, and the cobalt trichloro ion CoCl3 − (species with x = 3) controls the electrolytic properties [19, 29]. The large ΔE p difference between the anodic and cathodic peaks corresponding to Co2+/Co couple is comparable to the ΔE p values reported in the above-cited works. Nevertheless, the CVs recorded at slow scan rates (5–10 mV s−1) have a symmetric shape with approximately equal cathodic and anodic peak current. It might be considered that the Co2+/Co couple is a quasi-reversible process. The electrochemical behaviour of the couple at high scan rate changes completely. In this case, the obtained i pc /i pa ratio appears to be much greater than unity, demonstrating a higher degree of irreversible behaviour. Also, the gradual shift of peak potentials with increasing scan rate suggests a quasi-reversible process, although this shift can be due partially to a high uncompensated ohmic drop in ionic liquid media. This process is not a totally irreversible process because the reverse reaction of the electrode process occurs.
In order to demonstrate the mass-transport control of cathodic process, we represented the cathodic peak current density versus the square root of scan rate; straight lines passing approximately through the origin (not shown) for both Pt and GC electrodes were drawn for various Co2+ concentrations. For determining the diffusion coefficient (D) of Co2+, we used the well-known equations for an irreversible process [36]:
where I p is the cathodic peak current; n, number of electrons transferred; A, surface area of working electrode; C 0, CoCl2 molar concentration; α, the charge transfer coefficient; n α , number of electrons exchanged in the rate determining step; E p, the cathodic peak potential and E p/2, the cathodic half-peak potential; the other symbols have their usual meanings.
Table 1 shows the data of diffusion coefficients obtained taking n = 2, R = 8.314 J mol−1 K−1, T = 353.15 K and αn α values in the scan rate domain (50–200 mV s−1) where the process is irreversible.
The determined averaged value for D Co2+ diffusion coefficient using Pt electrode at 80 °C was 4.76 × 10−7 cm2 s−1, while for a GC electrode the value was 6.57 × 10−7 cm2 s−1. One should take into account that even the D Co2+ values obtained in this work varied with increasing cobalt ion concentration, however, they are in good agreement with the D Co2+ value (1.7 × 10−6 cm2 s−1 at 100 °C using tungsten electrode) reported by Li et al. in the same electrolyte [29]. The differences may be due to different temperature and electrode nature, as well as possible different structure of chloro complex cobalt ion present in the electrolyte. Also, these values obtained in our study are of a similar order of magnitude to the values of D Co2+ determined from some aprotic ionic liquids, such as butyl-methylimidazolium tetrafluoborate (0.18 × 10−7 cm2 s−1) [18] or butyl-methylpyrrolidinum bis-trifluoromethyl sulfonyl amide (1.00 × 10−7 cm2 s−1) [37]. As it is expected, the diffusion coefficient of Co2+ in ChCl-urea ionic liquids is with around two orders of magnitude lower than the diffusion coefficients reported in CoCl2 aqueous solutions: 0.53 × 10−5 [38], 1.10 × 10−5 [39] or 1.301 × 10−5 cm2 s−1 [40]. The explanation is related to the special physical-chemical properties of ionic liquid (viscosity, surface tension) that are very different than that for aqueous media.
Figure 2a–d show some examples of CV curves recorded on platinum and glassy carbon electrodes in ChCl-ethylene glycol (1:2) + CoCl2. In this system, the working temperature was established between 50 and 70 °C owing to a better fluidity of ionic liquid. One may see that the presence of the Co ions in solution gives rise to a cathodic plateau or a shoulder attributed to the reduction of Co(II) to metallic cobalt. The onset of cathodic current is within the potential range from −1 to −1.2 V vs. Ag. In general, the potential domain of the electroreduction process in the ChCl-EG + CoCl2 system is placed on more negative values compared to the ChCl-urea + CoCl2 system, suggesting a higher degree of irreversibility than in ChCl-urea, with mixed, diffusion and kinetic, control. Moreover, a broad anodic peak appears distinctly on CV curves from concentrated solutions (0.1 M Co2+). The increased irreversibility of Co2+/Co couple in ChCl-EG + CoCl2 system explains the low deposition rate of cobalt leading certainly to a less intense stripping process in anodic scan.

CVs in ChCl-EG with 0.01 M CoCl2 (a, b) and 0.1 M CoCl2 (c, d) at 70 °C, for Pt (a, c) and GC (b, d) electrodes
Although the viscosity (and conductivity) of the ChCl-EG system is clearly lower than that of the ChCl-urea system, the Co deposition seems to be more complicated in ChCl-EG. This may be due to the existence of a more stable Co complex ion in the ChCl-EG system (high overvoltage, low charge transfer). Perhaps a more distinct adsorption of chloro complex cobalt ions takes place at the electrode/electrolyte interface in the electrolyte with ChCl-EG solvent.
Figures 3 and 4 show the voltammograms recorded in ChCl-malonic acid (1:1) and ChCl-oxalic acid (1:1) mixtures with CoCl2 · 6H2O content. In the cathodic scanning domain, the shoulder or peak for cobalt deposition in these systems was not always well evidenced, in most cases CV curves showing only a continuous increase of cathodic current. We selected, however, some representative CV curves exhibiting clearly cathodic peaks or shoulders to demonstrate the occurrence of the cobalt deposition process on Pt or GC electrode. In general, the cathodic shape of CVs recorded in both ChCl-MA + CoCl2 and ChCl-OxA + CoCl2 systems suggests that the reduction process of Co2+ is hindered primarily by reduction of protons and the cathodic current may be the result of two contributions. Indeed, these electrolytes may have a high content of H+ ions provided not only from water molecules of cobalt hydrate salt but also from dissociation of free carboxilic acid molecules, which probably exist because of a disruption of hydrogen bonds. An excess of protons is expected in ChCl-OxA solvent which contains an excess of water, introduced supplementary as dihydrate oxalic acid. It should be mentioned that the metallic cobalt grains appearing on the surface of the electrode may serve as reaction sites for hydrogen evolution [2] and, therefore, the current for cobalt electrodeposition becomes indistinguishable from hydrogen evolution reaction. As a consequence, the reduction process from Co2+ to Co is not clearly observed on the cathodic branch of cyclovoltammograms. Moreover, on anodic branch of CVs, a peak corresponding to the oxidation of cobalt deposit does not occur very clearly even if the anodic scan is extended to +1.2 V vs. Ag (with Pt electrode) or to +1.4 V vs. Ag (with GC electrode), confirming the high irreversibility of the Co2+/Co process in the mixtures of choline chloride with carboxilic acids. All our findings demonstrate that the cobalt deposition process is not favoured from these acidic media.

CVs in ChCl-MA + 0.05 M CoCl2 electrolyte at 50 °C, on Pt (a) and GC (b) electrodes

CVs in ChCl-OxA + 0.5 M CoCl2 electrolyte at 60 °C, on Pt (a) and GC (b) electrodes
It is worth mentioning that the coordination of cobalt in CoCl2.6H2O-containing ionic liquids is complex and not fully understood, because a variety of complex species could be present. The results in the four basic electrolytes show that the Co chloro complex ions may be influenced differently by interaction with hydrogen bond donor molecules (urea, ethylene glycol, malonic acid or oxalic acid). The participation of water as a ligand in the coordination shell of cobalt should not be excluded, especially in ChCl-oxalic acid electrolyte.
Electrochemical impedance spectroscopy measurements
EIS measurements were performed in non-stirred electrolytes using the same four ionic liquid analogues and platinum or glassy carbon electrodes, to provide an insight into the reduction of Co2+ process occurring at the electrode/electrolyte interface. This investigation of the electrochemical behaviour of cobalt ion completes the information obtained from cyclic voltammetry. EIS curves were plotted considering four potential domains for polarization of the electrode towards negative potentials: before the onset of cobalt ion reduction, at this onset, at the potential where a peak was recorded in CV curves, and, finally, at very negative potentials where the choline ion reduction is involved, too. Some representative EIS plots are shown in Figs. 5, 6 and 7 as Nyquist diagrams.

Nyquist spectra for Pt (a) and GC (b) in ChCl-urea + 0.7 M CoCl2 electrolyte, 70 °C

Nyquist spectra for Pt (a) and GC (b) in ChCl-EG + 0.25 M CoCl2 electrolyte, 60 °C

Nyquist spectra for ChCl-carboxilic acid electrolytes. a ChCl-MA + 0.02 M CoCl2, GC electrode, 80 °C. b ChCl-OxA+ 0.25 M CoCl2, Pt electrode, 60 °C
The impedance spectra from ChCl-urea + CoCl2 and ChCl-EG + CoCl2 systems (Figs. 5 and 6) recorded at potentials before the onset of cobalt ion reduction (from −0.4 to −0.6 V vs. Ag) show a very large open capacitive loop (see complete curves in the insets). The response of the system is completely different at −0.7 V vs. Ag polarization. Here, the Nyquist plot within the high frequency range is a semicircle continued with a linear tail at low frequencies. The semicircular region corresponds to the electron transfer-limited process, in which the semicircle diameter is equal to the charge transfer resistance. The sloping straight line suggests the formation of Co film and, therefore, a solid state diffusion of Co2+ ions within the metallic film. The response of the system is similar for more negative polarizations of electrode, with semicircles having gradually smaller diameter, meaning a decrease in charge transfer resistance, i.e., enhanced electrolysis current.
The same single capacitive time constant, as open loop or full semicircle, is recorded in the representation of Nyquist spectra from ChCl-MA + CoCl2 and ChCl-OxA + CoCl2 electrolytes (Fig. 7). Especially in these systems, at all electrode polarizations of the electrode, the semicircle is depressed, indicating a complex nature of the surface which has probably a high roughness or surface defects. At lower frequencies, a dispersion of the impedance values is observed. This supports the above assumption of an increased irreversibility for reduction of Co2+ taking place in the binary mixtures of choline chloride with carboxylic acids. The appearance of an inductive portion within the loop at low frequencies, especially in the ChCl-OxA system (Fig. 7b), is probably associated with the adsorption of cobalt adatoms [19]. Also, the existence of adsorbed hydrogen molecules may be possible.
In EIS spectra recorded from all four electrolytes, the thickening of Co film also leads to changes in the values of the maximum phase angle and impedance modulus, these parameters belonging to Bode diagrams (not shown). Accordingly, the corresponding Bode plots show a decrease of maximum phase angle from −60° on Pt or −70° on GC, to values of −20° up to −5°. The behaviour might be explained by an increase in electrical conductivity for thicker film, although probably the deposit is not a pure metal.
The obtained EIS spectra were interpreted on the basis of equivalent electrical circuits as electrochemical models of the interface, using specialized fitting software ZView 2.90 (Scribner Assoc.). Figure 8 exhibits the proposed equivalent circuits. One is the Randles circuit (Fig. 8a) which includes the solution resistance (R1), a constant phase element (CPE1, instead of double-layer capacitance, C dl) in parallel with a charge transfer resistance (R2). The second model (Fig. 8b) is a more complex circuit and consists of the following components: a solution resistance (R1); a constant phase element (CPE1) in parallel with an ohmic resistance (R2), both attributed to film capacitance and film resistance, respectively; additional parallel connection between CPE2 (instead of double-layer capacitance); and the charge transfer resistance for Co2+ reduction process (R3). The impedance of CPEs is given by the 1/T(jω)p ratio, where \( j=\sqrt{-1} \), ω is the angular frequency, T is a constant in F cm−2 sp-1 that signifies the capacitive behaviour and p is the the exponent related to the angle of the impedance vector on the complex plane plots; only when p = 1, T ≡ C dl and purely capacitive behaviour is obtained.

The proposed equivalent circuits used for data fitting. a For ChCl-OxA+ 0.25 M CoCl2. b For ChCl-urea + 0.7 M CoCl2, ChCl-EG + 0.25 M CoCl2 and ChCl-MA + 0.02 M CoCl2
The proposed equivalent circuit (a) fitted better the experimental impedance data for choline chloride-oxalic acid system. The model cell (b) was fitted to the impedance measurements performed in binary mixtures of choline chloride with urea, ethylene glycol or malonic acid. The values of the equivalent circuit elements for the best fitting of experimental data were calculated. The obtained ohmic resistances of the solutions have, as expected, similar values (50–60 Ω cm2), except the case of ChCl-urea (ca. 120 Ω cm2). The values of the same order of magnitude for the components associated with the Co film formed were determined in various polarization states with CPE1-P values from circuit (b) being close to unity (0.8–1). The double-layer capacitance approximated as the corresponding CPE-T component was equal to 150-300 × 10−6 F cm−2 sp-1 (with p also close to unity). This is a reasonable value, taking into account some surface roughness and higher viscosity of ionic liquids than of aqueous solutions. The values of the charge transfer resistance obtained by fitting confirm the values of Nyquist semicircle diameters. They decrease gradually by shifting the electrode potential towards negative direction.
Electrodeposition and characterization of cobalt thin films
Potentiostatic electrolysis was carried out for 1 h at 60 °C to deposit cobalt films on copper substrate from all four employed ionic liquid analogues with identical Co2+ concentration (0.5 M CoCl2). Representative samples of Co films were obtained by applying potentials of −0.9 V vs. Ag for ChCl-urea eutectic ionic liquid (sample I) and −1.1 V vs. Ag for the other solvent media: ChCl-EG (sample II), ChCl-MA (sample III) and ChCl-OxA (sample IV). A single experiment with Co coating on copper substrate (sample V) was performed in ChCl-EG + 0.5 M CoCl2 system by carrying galvanostatic electrolysis for 1 h at 70 °C, with 5 mA cm−2 current density, parameters selected from Hull cell experiments. The electrolyses have been performed in naturally aerated and stationary conditions. The experimental film thickness values (around 0.5 μm) were close to the theoretical thickness based on Faraday’s electrolysis law. Taking into account this thickness, we did not expect these thin films to have very good magnetic or microhardness performances, or high corrosion resistance.
The appearance of the best Co films was grey in colour, as smooth, adherent and uniform deposits, but with a rough surface leading to an optical dark grey shade. In some additional tests made at more negative potentials, the deposits had an aspect of black powder coating, and the cobalt particles were loose from the substrate.
Figure 9 show SEM images and EDX spectra for Co films prepared from ChCl-urea (sample I, Fig. 9a, b) and from ChCl-EG (sample II, Fig. 9c, d) in potentiostatic conditions. The SEM micrographs illustrate a change in the morphology of films: sample I is a more uniform film, having smaller granular particles when compared with sample II deposited under quite similar conditions. This may be a good demonstration that in ChCl-EG, the nucleation rate is low and the growth rate is high. Figure 9a, c also indicates the five selected places of each sample where EDX elemental analysis was performed. The elemental composition data of Co films are listed in Table 2. The data analysis shows that the film prepared from ChCl-urea consists essentially of cobalt, 92–94 wt%, with small amounts of chlorine and oxygen, due to traces of ionic liquid electrolyte retained inside the film. The cobalt content in sample II is much smaller, 52–67 wt% Co, with relatively high concentrations of oxygen and chlorine (35 and 5 wt%, respectively).

SEM micrographs and EDX elemental analysis spectra for sample I deposited from ChCl-urea (a, b) and sample II deposited from ChCl-EG (c, d) confirming the cobalt content in deposited films
Cobalt films from ChCl-MA (sample III) and ChCl-OxA (sample IV) have an appearance of a dark grey deposit. The powdery nature of such deposits is expected, as they are produced under conditions where Co2+ is reduced together with hydrogen at a high rate of evolution. SEM micrographs for these films are shown in Fig. 10. The effect of hydrogen evolution dominance leading to a very thin and felt-like film becomes clearly evident on the coating produced from ChCl-OxA (sample IV). EDX analysis data for samples III and IV showed a very low cobalt content (around 30 and 15 wt%, respectively) in the deposits.

SEM micrographs of Co thin films deposited from ChCl-MA and ChCl-OxA ionic liquids with 0.5 M CoCl2 as precursor, 60 °C. a Sample III (from ChCl-MA). b Sample IV (from ChCl-OxA)
It was seen that the Co film does not change very much its morphology with the deposition time. Samples I and II are different in surface morphology. Sample I is granular and relatively rough; sample II is granular, less compact and porous-like. In addition, sample II contains more impurities in the Co film. It is obvious that the compact morphology of sample I might prevent the aggressive ion (chloride ion) penetration in an enhanced manner when compared to the cases of samples II–IV. A change of electrolysis conditions from a constant potential deposition (potentiostatic) to the constant current deposition (galvanostatic) does not have a strong effect on the microstructure of the films (see the SEM micrograph in Fig. 11a). The film deposited with current control (sample V) has the polyhedral irregular aggregates particles, with visible crystallographic facets.

SEM micrograph (a) and XRD pattern (b) for Co film prepared from ChCl-EG + 0.5 M CoCl2, 5 mA cm−2 controlled current density, 60 min, 70 °C
Figure 11b shows a typical X-ray diffraction (XRD) pattern for as-deposited Co films. The XRD spectrum displays two peaks at 2θ angle equal to 48.8 and 55.8°, which are characteristic of (100) and (101) diffraction plans. As a result, the structural analysis using powder XRD diffraction studies has indicated the existence of a multilayer hexagonal centred (hcp) cobalt structure. This result is similar to the hcp structure of Co films obtained by Suzaki and Watanabe [9] at low current density in organic solvent baths. However, the additional data resulting from detected signals centred at 2θ around 52 and 61° indicate the additional presence of face-centred-cubic (fcc) cobalt. The Cu substrate characteristic peaks are also evidenced at 50.3 and 58.9°, suggesting a Co deposit as a very thin film.
Figure 12a–c shows VSM images determined with vibrating sample magnetometer exhibiting the behaviour of as-deposited Co films (deposits of 5 mg with 0.5 μm thickness) in the presence of an external magnetic field. The mass magnetization σ (denoted also as specific magnetic moment) increases with magnetizing field strength (H), reaching saturation in both positive and negative directions. The shape of magnetization curves is typical for ferromagnetic materials. The experimental magnetic data are listed in Table 3 and are in good agreement with the finding of other authors [19] regarding Co thin films. The data dispersion is related to the influence of the operating conditions during film deposition (controlled electrode potential or current density, chemical nature of the bath and temperature). The values of remanence show that all Co films are able to be magnetized. When driving magnetic field drops to zero, the Co film retained a considerable degree of magnetization, especially for sample II. The coercivity (coercive force) is an intercept of the hysteresis loop (the applied field) where the data line crosses zero and illustrates a relatively high resistance of ferromagnetic material to becoming demagnetized. Therefore, the obtained Co films may be used as magnetically soft materials [41]. It was noted that the film prepared from ChCl-urea + 0.5 M CoCl2 electrolyte (sample I) yielded the highest in-plane coercive field. This demonstrates an ordered magnetic phase that has a larger contribution compared to the other magnetic films (samples II or V). Increased value of coercivity in this case may indicate a significant stress present inside the deposit, because both remanence and saturation of sample I are lower than for sample II.

Hysteresis loops of Co films on applying the magnetic field. a Parallel (in-plane) position of samples I and II. b, c Parallel (angle 0°) and perpendicular-to-plane (angle 90°) positions of sample V
A magnetic anisotropy of sample V was emphasized by plotting hysteresis loops at two different angles with the external magnetic field: 0° (parallel, in-plan direction) and 90° (perpendicular-to-plane direction), respectively. This anisotropy (shown in Fig. 12b, c) may be due to a special texture of Co deposit. The coercive force in-plane position of this film is about 100 Oe which is more than two times greater than the coercive force in a perpendicular position. Also, the hysteresis loops have not been centred at zero point, therefore the magnetic properties of sample V in Table 3 had to be approximated.
Conclusions
The investigations by using cyclic voltammetry and electrochemical impedance spectroscopy showed the possibility to evidence the electrochemical behaviour of Co2+ in all four ionic liquid analogues based on choline chloride. Cyclic voltammograms of Co2+ ion in the eutectic solvents of ChCl mixed with urea or ethylene glycol show a cathodic peak of direct reduction to Co metal and an anodic peak related to stripping/dissolution of Co deposit from Pt or glassy carbon electrode. The linear dependencies of peak current vs. square scan rate suggest a diffusion control. However, the process is quasi-reversible at slow scan rates and behaves irreversible at high scan rates. The calculated values of the diffusion coefficients of Co2+ in ChCl-urea at 80 °C were 4.76 × 10−7 cm2 s−1 (on Pt) and 6.57 × 10−7 cm2 s−1 (on GC), respectively, with two orders of magnitude smaller than for aqueous solutions. The cathodic reduction of Co2+ was not clearly evidenced in CVs in eutectic mixtures of ChCl with malonic acid or with oxalic acid, due to a high degree of irreversibility and simultaneous hydrogen evolution.
Nyquist impedance spectra evidenced the deposition of cobalt metal: the capacitive semicircle from high frequencies is continued with a straight line at low frequencies, suggesting the formation of Co film and a barrier for diffusion of Co2+ ions within the metallic film. Correspondingly, both maximum phase angle and impedance modulus in the Bode plots decrease gradually with the increase of cathodic polarization of the electrode.
Co films were electrodeposited effectively onto the copper surface. SEM micrographs showed different appearance and morphology, depending on the chemical nature of ionic liquid analogue used as bath. Grey in colour, smooth, adherent and uniform Co deposits were obtained from ChCl-urea and ChCl-EG mixtures. The structural constitution evidenced by X-ray diffraction indicated a multilayer hexagonal centred (hcp) crystalinity. Dark grey and felt-like or even powdery films were obtained from ChCl-MA and ChCl-OxA solvents. EDX elemental analysis proved that Co films with higher cobalt content are obtained from ChCl-urea (92–94 wt% Co), while films from ChCl-ethylene glycol have a lower content in metallic cobalt (52–67 wt% Co). However, both kinds of Co films behave as typical ferromagnetic material.
References
Mendoza-Huizar LH, Rios-Reyes CH (2011) J Solid State Electrochem 15(4):737–745
Mendoza-Huizar LH, Rios-Reyes CH (2012) J Solid State Electrochem 16(9):2899–2906
Mendoza-Huizar LH, Rios-Reyes CH (2013) Cent Eur J Chem 11(8):1381–1392
Rios-Reyes CH, Mendoza-Huizar LH, Reyes-Cruz VE, Rodriguez MAV (2013) Quim Nova 36(7):978–983
Prasad KA, Giridhar P, Ravidran V, Muralidharan VS (2001) J Solid State Electrochem 6:63–68
Krastev I, Dobrovolska T, Lacnjevac U, Nineva S (2012) J Solid State Electrochem 16:3449–3456
Zhou QF, Lu LY, Yu LN, Xu XG, Jiang Y (2013) Electrochim Acta 106:258–263
Cui CQ, Jiang SP, Tseung ACC (1991) J Electrochem Soc 138:94–100
Suzaki A, Watanabe T (2000) J Jpn Inst Metals 64(10):869–877
Yamamoto H, Morishita M, Mizuta Y, Masubuchi A (2012) Surf Coat Technol 206:3415–3420
Carlin RT, De Long HC, Fuller J, Trulove PC (1998) J Electrochem Soc 145:1598–1607
Chen P-Y, Sun I-W (2001) Electrochim Acta 46:1169–1177
An MZ, Yang PX, Su CN, Nishikata A, Tsuru T (2008) Chin J Chem 26(7):1219–1224
Freyland W, Zell CA, El Abedin SZ, Endres F (2003) Electrochim Acta 48(20–22):3053–3061
Lin LG, Yan JW, Wang Y, Fu YC, Mao BW (2006) J Exp Nanosci 1(3):269–278
Schaltin S, Nockeman P, Thijs B, Binnemans K, Fransaer J (2007) Electrochem Solid State Lett 10:D104–D107
Yang PX, An MZ, Su CN, Wang FP (2008) Electrochim Acta 54:763–767
Su CN, An MZ, Yang PX, Gu HW, Guo XH (2010) Appl Surf Sci 256:4888–4893
Ali MR, Nishikata A, Tsuru T (2005) Indian J Chem Technol 12:648–653
Katayama Y, Fukui R, Miura T (2007) J Electrochem Soc 154(10):D534–D537
Katayama Y, Fukui R, Miura T (2007) ECS Trans 3:287–295
Fukui R, Katayama Y, Miura T (2011) Electrochim Acta 56:1190–1196
Ispas A, Buschbeck M, Pitula S, Mudring A, Uhlemann M, Bund A, Endres F (2009) ECS Trans 16(45):119–127
Abbott AP, Davies DL, Capper G, Rasheed RK, Tambyrajah V (2004) Ionic liquids and their use as solvents, US Patent 2004/0097755 A1 (May 20, 2004)
Yue D, Jia Y, Yao Y, Sun J, Jing Y (2012) Electrochim Acta 65:30–36
Gomez E, Cojocaru P, Magagnin L, Valles E (2011) J Electroanal Chem 658:18–24
Cojocaru P, Magagnin L, Gomez E, Valles E (2011) Mater Lett 65:3597–3600
Guillamat P, Cortes M, Valles E, Gomez E (2012) Surf Coat Technol 206:4439–4448
Li M, Wang Z, Reddy RG (2014) Electrochim Acta 123(20):325–331
Abbott AP, Capper G, Davies DL, Rasheed R (2004) Chem Eur J 10:3769–3774
Abbott AP, Davies GL, Capper G, Rasheed RK, Tambyrajah V (2007) Ionic liquids and their use, US Patent No. 7,196,22 (March 27, 2007)
Srivastava M, Yoganandan G, William Grips VK (2012) Surf Eng 28(6):424–429
You YH, Gu CD, Wang XL, Tu JP (2012) Surf Coat Technol 206:3632–3638
Ciocirlan O, Iulian O, Croitoru O (2010) Rev Chim (Bucharest) 61(8):721–723
Mares (Badea) ML, PhD Thesis, Univ. POLITEHNICA of Bucharest, 2013
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications, 2nd edn. John Wiley, New York
Fukui R, Katayama Y, Miura T (2005) Electrochemistry 73:567–569
Cui CQ, Jiang SP, Tseung ACC (1990) J Electrochem Soc 137(11):3418–3423
Ribeiro ACF, Lobo VMM, Natividade JJS (2002) J Chem Eng Data 47:539–541
Ribeiro ACF, Valente AJM, Costa DO, Simoes SMN, Pereira RFP, Lobo VMM, Esteso MA (2010) Electrochim Acta 55(15):4483–4487
Kaye & Laby Tables of physical and chemical constants, chapter 2.6.6 Magnetic properties of materials, NPL (National Physical Laboratory USA), 19th edition, 1995
Acknowledgments
One of the authors (M.L. Mares) recognizes the financial support from the European Social Fund through POSDRU/107/1.5/S/76813 Romanian Research Project. Part of this work was financially supported under M ERA Net Program, NANOCOATIL 7-082/2013 Research Project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cojocaru, A., Mares, M.L., Prioteasa, P. et al. Study of electrode processes and deposition of cobalt thin films from ionic liquid analogues based on choline chloride. J Solid State Electrochem 19, 1001–1014 (2015). https://doi.org/10.1007/s10008-014-2711-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-014-2711-9