
Abstract
In this work, a modified carbon paste electrode consisting of Nickel dispersed in poly(ortho-aminophenol) was used for electrocatalytic oxidation of methanol in alkaline solution. A carbon paste electrode bulk modified with o-aminophenol was used for polymer preparation by cyclic voltammetry method; then, Ni(II) ions were incorporated by immersion of the modified electrode in 1 M Ni(II) ion solution at open circuit. The electrochemical characterization of this modified electrode exhibits stable redox behavior of the Ni(III)–Ni(II) couple. Electrocatalytic oxidation of methanol on the surface of modified electrode was investigated with cyclic voltammetry and chronoamperometry methods, and the dependence of the oxidation current and shape of cyclic voltammograms on methanol concentration and scan rate were discussed. Also, long-term stability of modified electrode for electrocatalytic oxidation of methanol was investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
For the last decade, methanol has been considered to be a liquid fuel of relatively high activity in fuel cell systems [1–5]. Considerable efforts have been directed towards the study of methanol electrooxidation in solutions of a high pH [6–9]. The use of alkaline solutions in a fuel cell has many advantages such as increasing its efficiency [6, 10–11], a wider selection of possible electrode materials, a better efficiency of oxygen cathode, and oxidation reactions of organic fuels exhibit almost no sensitivity to the surface structure [12]. It was found that the graphite-supported platinum electrodes exhibited almost the same specific activity as a smooth Pt electrode for methanol oxidation in NaOH solution [13]. Smaller or negligible poisoning effects in alkaline solutions were observed [14]. In the electrochemical oxidation of methanol, the electrode material is clearly an important parameter where a high efficient electrocatalyst is needed. The mechanism and kinetics of methanol oxidation have been studied under a wide range of solution conditions and at several electrodes including Pt, Pt oxides [15–21], Pt-Sn [22], Pt-Ru [23–25], and nickel [26–31].
Although metals such as Pt and Pt-Ru are very active in the anodic oxidation of methanol, they are too expensive for practical applications, and unfortunately, compounds such as methanol with large oxidation overpotential at ordinary carbon electrodes are not suitable analytes for these electrodes. One promising approach for minimizing overvoltage effects is in carrying out the electrocatalytic process at chemically modified electrodes. In this context, during the last years, a large number of papers dedicated to the electrocatalytic properties of metallic microparticles incorporated into polymeric matrices have been published [32–36]. Among a number of polymeric metal complexes, those containing Ni(II)–Ni(III) redox couple have received considerable attention in recent years [37–39]. This is partly due to the fact that their behavior in alkaline solution is reminiscent of a nickel oxyhydride species which is commonly believed to act as a redox mediator between a substrate and an electrode in many electrooxidaton processes. Maximovitch and Bronoel [40] reported that it is difficult to measure the oxidation current of methanol on smooth nickel because it is difficult to obtain a surface free from adsorbed oxygen. Smooth nickel is very sensitive to molecular oxygen dissolved in the solution that adsorbs on the electrode and inhibits the reaction.
In the previous work, we used the poly(1-naphtylamine)/Ni-modified carbon paste electrode for electrocatalytic oxidation of several carbohydrates [41]. This study showed that the metal-polymer electrodes are easy to prepare, stable for long time periods with good detection limits, and have wide linear range responses. Also, recently, we have demonstrated that the poly-1,5-Diaminonaphthalene/Ni-modified carbon paste electrode can successfully catalyze the oxidation of methanol in alkaline medium [42]. In these studies, we have combined the advantageous features of polymer modification, dispersion of metallic particles into an organic polymer, and carbon paste technology. All these results encouraged us to continue the studies with new polymer materials.
In this work, we decided to combine the above-mentioned advantageous features again for the aim of electrocatalytic oxidation of methanol by use of the poly(o-aminophenol) as a new polymer. We introduced o-aminophenol monomer into the carbon paste bulk and obtained a polymeric-coated electrode in situ by electropolymerization of the monomer. In fact, this work showed that a renewable and reproducible polymeric-coated electrode could be generated rapidly at the o-aminophenol-modified carbon paste electrode (OAP/MCPE). Then, nickel ions were incorporated into the polymeric matrix by immersion of polymeric-modified electrode in a nickel nitrate solution. This electrode was used for the electrocatalytic oxidation of methanol in NaOH solution.
Experimental
Reagent and materials
OAP from Merck was recrystallized in ethyl acetate and dried under vacuum. Colorless OAP crystals were protected from light in a dark desiccator [43]. Perchloric acid from Fluka was used as the supporting electrolyte. All other reagents were of analytical grade.
Instrumentation
Electrochemical experiments were performed on 746VA Trace Analyzer Metrohm potentiostat with a Metrohm voltammetry cell in a three-electrode configuration. An Ag–AgCl was used as reference electrode, and a platinum wire was the auxiliary electrode. Working electrode was Nickel/poly(o-aminophenol)-modified carbon paste electrode (Ni/POAP/MCPE).
Working electrode
A 1% (w/w) mixture of OAP to total weight of graphite powder was made by dissolving a given quantity of OAP in diethyl ether and hand mixing with the required graphite powder in a mortar and pestle. The solvent was evaporated by stirring. Silicon oil was then added and was blended by hand mixing until a homogeneous paste was obtained. For fabrication of the electrode, the prepared paste was tightly packed into one end of a glass tube (approximately 3.4 mm internal diameter), and a copper wire was introduced into the other end for electrical contact. A fresh electrode surface was generated rapidly by extruding a small plug of the paste with a stainless steel rod and smoothing the resulting surface on white paper until a smooth shiny surface was observed.
Result and discussion
Preparation of poly(o-aminophenol)-modified carbon paste electrode
Previously, poly(o-aminophenol) films were obtained at the surface of Pt and GC electrodes by use of cyclic voltammetric method [43, 44]. In this work, we investigated the preparation of poly(o-aminophenol) in the bulk of carbon paste electrodes. Electropolymerization was carried out at the OAP/MCPE by cycling the potential between 0 to 750 mV in 0.5 M HClO4 solution. Figure 1 shows typical multisweep cyclic voltammograms of modified electrode during electropolymerization. As can be seen from this figure, OAP oxidizes irreversibly at 700 mV in the first positive scan without corresponding cathodic processes in the reverse scan. The second and consecutive scans are different; the reaction begins at higher potential and two redox peaks (related to polymer) in lower potentials appeared, and their currents increased with potential cycling. The shape of voltammograms obtained during the polymerization on the surface of this electrode is not exactly corresponding to those reported in the literature [43, 44], because the monomer is in the bulk of carbon paste; therefore, its reaching to electrode surface is easy and blocking effect is not a problem.

Consecutive cyclic voltammograms of OAP/MCPE in 0.5 M HClO4 solution scan rate 100 mV s−1
After preparation of poly(o-aminophenol), the electrode was removed, rinsed with water, and the sides wiped with soft tissue paper. The redox behavior of poly(o-aminophenol) was investigated in the electrolyte solution (0.5 M HClO4 with scan rate of 20 mV s−1). Cyclic voltammograms of freshly prepared electrode in 0.5 M HClO4 shows two redox couple at potentials about 0.15 and 0.35 V (Fig. 2a), but after immersing the electrode for 1.5 h in 0.5 M HClO4 solution, the redox couple in 0.35 V disappeared (Fig. 2b). Therefore, this redox couple can be attributed to the soluble product of 2-aminophenoxazin-3-one (APZ; Scheme 1(A)) which can be produced during electropolymerization behind the main structure of POAP films (phenoxazine units Scheme 1(B)) [43] and because of its solubility that can diffuse from the carbon paste bulk to the electrolyte solution.

Electrochemical responses of POAP/MCPE: a immediately, b after 1.5 h remaining in 0.5 M HClO4 solution, c in 0.1 M NaOH solution. scan rate 20 mV s−1

Structure of A APZ and B phenoxazine units
The response obtained in a 0.1-M NaOH solution showed a complete loss of electrode activity in the potential range from 0 to 0.75 V (Fig. 2c). However, the film was not degraded under these experimental conditions, and its response was recovered when the electrode was immersed in a supporting electrolyte solution of HClO4 0.5 M.
Incorporation of Ni(II) ions into poly(o-aminophenol) film and electrochemical response of the Ni/POAP/MCPE
In order to incorporate Ni(II) ions into the POAP film, the freshly electropolymerized OAP/MCPE was placed at open circuit in a well-stirred aqueous solution of 1 M Ni(NO3)2. Accumulation of nickel was carried out by complex formation between Ni(II) in solution and amines sites in the polymer backbone [45, 46] for a given period of time (t a, accumulation time). The polarization behavior was examined in 0.1 M NaOH for Ni/POAP/MCPE using cyclic voltammetry (CV). This technique allows the oxide film formation in parallel to inspecting the electrochemical reactivity of the surface [47]. Voltammograms were recorded by cycling the potential between 0.1 and 0.85 V at 100 mV s−1 until a stable voltammograms was obtained. Figure 3 shows the electrochemical response of the POAP/MCPE and Ni/POAP/MCPE after polarization in 0.1 M NaOH solution. From Fig. 3, it can be seen that whereas neither oxidation nor reduction took place on the POAP/CPE, a well-developed redox wave was observed on the Ni/POAP/MCPE when the potential was cycled between 0 and 0.85 V, which was related to the oxidation of Ni(OH)2 to NiOOH with a peak potential of 0.51 V and reduction of NiOOH to Ni(OH)2 with a peak potential of 0.31 V. The surface coverage of the immobilized active substance (Ni(II)) in the films can be evaluated from the charge under the current–potential wave (Fig. 3b) with correction for the baseline (Γ* = Q/nFA). The value of Γ* for Ni/POAP/MCPE was 1.37 × 10–5 mol cm−2.

Electrochemical responses of electrodes: a POAP/MCPE and b Ni/POAP/MCPE, in 0.1 M NaOH solution, scan rate 20 mV s−1
Figure 4A shows the cyclic voltammograms of Ni/POAP/MCPE in 0.1 M NaOH solution at different potential sweep rate in a wide range of 10–400 mV s−1; as can be seen. anodic and cathodic peak currents are linearly proportional to the potential sweep rate at low values from 10 to 50 mV s−1 (Fig. 4B). This can be attributed to an electrochemical activity of an immobilized redox couple at the surface. In the higher range of potential sweep rates (70–400 mV s−1; Fig. 4C), the peak currents depend on square root of the potential sweep rate, signifying the dominance of a diffusion process as the rate limiting step in the total redox transition of the modifier film. This limiting diffusion process was also reported for other Ni-modified electrodes [48, 49].

A Cyclic voltammograms of Ni/POAP/MCPE in 0.1 M NaOH solution. Potential sweep rates from inner to outer are: 10, 20, 25, 30, 50, 70, 100, 200, and 400 mV s−1, respectively. B The dependency of Current on v at lower values (10–50 mV s−1). C The dependency of Current on v 1/2 at higher values (70–400 mV s−1)
As has been reported [50–53], nickel hydroxide may generally exist in two different crystallographic forms designed α-Ni(OH)2 and β-Ni(OH)2 which are hydrous and anhydrous, respectively. In addition [53, 54], the oxidation of nickel hydroxide gives two other varieties of oxyhydroxides, β and γ, which explains the existence of the two reduction peaks during the backward sweep observed in high potential sweep rate in Fig. 4A.
Electrocatalytic oxidation of methanol at Ni/POAP/MCPE
Cyclic voltammetry studies
In this work, the electrochemical behavior of methanol was first studied at a POAP/MCPE electrode (without the incorporation of nickel) by cyclic voltammetric experiments in 0.1 M NaOH. Typical results obtained for a potential range of 0 to 1.2 V vs. Ag–AgCl are shown in Fig. 5A. The electrochemical response of POAP/MCPE in the absence of methanol is shown in Fig. 5A(a); the addition of 0.43 M methanol to the alkaline solution causes no effect on the electrochemical response of the POAP/MCPE (Fig. 5A(b)). The electrochemical response of a Ni/POAP/MCPE in alkaline solution (i.e., 0.1 M NaOH) exhibits well-defined anodic and cathodic peaks (Fig. 5B(a)) associated with the Ni(II)–Ni(III) redox couple. As can be seen, upon methanol addition (0.43 M), there is an increase in the anodic peak current and a decrease in the cathodic peak current (Fig. 5B(b)). This behavior is typical of that expected for mediated oxidation as follows:

Electrochemical responses of A POAP/MCPE and B Ni/POAP/MCPE to: a 0 and b 0.43 M methanol in 0.1 M NaOH solution, scan rate 20 mV s−1
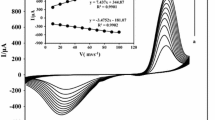
As can be seen in Fig. 5B, the oxidation current of methanol on the surface of Ni/POAP/MCPE appears at a potential slightly more positive than that of Ni(II)/Ni(III) oxidation. In fact, this peak is a new anodic peak. In order to further clarify the electrochemical oxidation mechanism of methanol on the Ni/POAP/MCPE, effect of methanol concentrations and potential scan rates on cyclic voltammetric responses was investigated. Figure 6A represents the CVs of the Ni/POAP/MCPE electrode in the range of 0–0.85 V at the scan rate of 20 mV s−1. The concentration of methanol in 0.1 M NaOH solution is changed from 0 to 2.4 M. It is observed from Fig. 6B that in low concentration of methanol, there are two anodic peak; a1 (assigned to the couple α-Ni(OH)2/NiOOH) and a2 (assigned to β-Ni(OH)2/NiOOH).

A Main panel: cyclic voltammograms of the Ni/POAP/MCPE in 0.1 M NaOH solution with different concentrations of methanol: a 0, b 0.04, c 0.15, d 0.22, e 0.43, f 0.66, g 0.99, h 1.38, i 2, and j 2.4 M, respectively. Inset: Plot of catalytic current vs. methanol concentration. B Zoomed voltammograms of b, c, and f from main panel of A
The corresponding electrode reaction involved in the anodic peak a2 might be
Therefore, the Ni(OH)2 produced in reaction 2 might be β-Ni(OH)2 [52]; as mentioned above, crystallographic form of this Ni(OH)2, represented by β, is different from that of the α-Ni(OH)2 involved in the anodic peak a1. This is consistent with different redox potentials of α-Ni(OH)2/NiOOH and β-Ni(OH)2/NiOOH [56], i.e., α-Ni(OH)2 is converted to NiOOH at a lower potential than β-Ni(OH)2 to NiOOH. It is observed from Fig. 6B that when methanol concentration increases, the current density of the peak a2 increases significantly while the peak a1 decreases and even disappears when the concentration of methanol becomes more than 0.22 M. This result shows that much more β-Ni(OH)2 is generated through reaction 2, and subsequently, the current density of the peak a2 increases considerably, as shown in Fig. 6.
Figure 7A represents the CVs of the Ni/POAP/MCPE electrode in the presence of 0.45 M methanol concentration at different potential scan rates. It is observed from Fig. 7 that in low potential scan rates, only peak a2 exists, and in potential scan rates higher than 100 mV s−1, peak a1 appears and its current density increases with potential scan rates increasing, while the peak a2 decreases and even disappears when the potential scan rates become more than 600 mV s−1.

A Main panel: cyclic voltammograms of the Ni/POAP/MCPE in the presence of 0.45 M methanol at different scan rates: a 10, b 20, c 30, d 50, e 70, f 100, g 200, h 300, i 400, and j 600 mV s−1, respectively. Inset: Variations of anodic current function vs. v. B Zoomed voltammograms of b, f, and i from main panel of A
These results, and also plotting the current function (anodic peak current divided by the square root of the potential sweep rate) against the potential sweep rate (inset of Fig. 7A), revealed negative slope confirming the electrocatalytic nature of the process.
The redox couple Ni(II)–Ni(III) has played a mediate role on heterogeneous catalytic-oxidation of methanol. In the redox system, α-Ni(OH)2 is firstly oxidized to NiOOH through the electrode reaction 1 in an alkaline medium; then, the generated NiOOH is reduced to β-Ni(OH)2 by methanol through reaction 2. Subsequently, this β-Ni(OH)2 is converted to NiOOH at higher potentials through reaction 3, leading to the appearance of a new anodic peak. Electrochemical oxidation processes of methanol on the Ni/POAP/MCPE can be exhibited in Scheme 2.

Schematic diagram of electrocatalytic oxidation of methanol on the surface of Ni/POAP/MCPE
The anodic currents for methanol electrooxidation under the same conditions at the surface of different electrodes are given in Fig. 8. As can be seen, conventional Pt electrode has negligible catalytic effect for methanol and also in the absence of polymer the amount of accumulated Ni (electrode (2)); therefore, its catalytic effect for methanol oxidation is very low. In electrode (3), the POAP was prepared on the surface of a bare carbon paste electrode, with electropolymerization in 5 mM monomer solution which, to some extent, improved methanol oxidation current. As can be seen in electrode (4), in which polymer prepared on the surface of monomer containing carbon paste electrode, the amount of accumulated Ni and catalytic current for methanol oxidation is significantly higher.

Comparison of anodic currents for various electrodes: (1) Pt in 1 M H2SO4 solution, (2) Ni/CPE, (3) Ni/POAP/CPE, and (4) Ni/POAP/MCPE in 0.1 M NaOH solution in the absence (checkered bar), and presence (filled bar) of 0.1 M methanol. Scan rate: 20 mV s−1
Chronoamperometric studies
Double potential step chronoamperometry, as well as other electrochemical methods, was employed for the investigation of electrochemical processes at Ni/POAP/MCPE. The main panel of Fig. 9 represents the current–time profiles obtained by setting the working electrode potential at 700 mV (in first step) and 350 mV (in second step) for various concentrations of methanol. The forward and backward potential step chronoamperometry of the modified electrode in the blank solution that showed an almost symmetrical chronoamperogram with almost equal charges consumed for the oxidation and reduction of surface confined Ni(II)/Ni(III) sites (Fig. 9A(a’)). However, in the presence of methanol, the charge value associated with the forward chronoamperometry, Q, is greater than that observed for the backward chronoamperometry (Fig. 9A(f’)).

Chronoamperograms obtained at the Ni/POAP/MCPE in absence (a) and presence of (b) 0.09, (c) 0.45, (d) 1, (e) 1.4 and (f) 2.3 M of methanol, first and second potential steps were 0.7 and 0.35 V vs. Ag–AgCl, respectively, in 0.1 M NaOH solution. Inset (A) dependence of Charge (µC) vs. t, a’ and f’, respectively, derived from the data of chronoamperograms of (a) and (f). Inset (B) dependence of I C/I L on t 1/2 derived from the data of chronoamperogams of (a) and (f) in the main panel. Inset (C) dependence of current (μA) on t −1/2 derived from the data of chronoamperogams of (a) in the main panel
The rate constant for the chemical reaction between the methanol and redox sites of Ni/POAP/MCPE can be evaluated by chronoamperometry according to the method described in the literature [57].
Where I C is the catalytic current of the Ni/POAP/MCPE in the presence of methanol, I L is the limiting current in the absence of methanol, and γ = kc o t (co is the bulk concentration of methanol) is the argument of the error function. In the cases where γ exceeds 2, the error function is almost equal to 1 and the above equation can be reduced to:
Where k, c o, and t are the catalytic rate constant (cubic centimeter per mole per second), methanol concentration (mole per cubic centimeter), and time elapsed (seconds), respectively. From the slope of the I C/I L vs. t 1/2 plot, we can simply calculate the value of k for a given concentration of substrate. Inset (B) of Fig. 9 shows one such plot, constructed from the chronoamperogram of the Ni/POAP/MCPE in the absence and presence of 2.3 M methanol. The mean value for k was found to be 86.7 cm3 mol−1 s−1. Inset (C) of Fig. 9 shows the cotrel curve for modified electrode obtained from chronoamperogram (a); the linear relationship amplifies the diffusion controlled behavior of nickel in polymeric matrix.
Stability of Ni/POAP/MCPE
We checked long-term stability of Ni/POAP/MCPE by measuring its response for methanol oxidation after 1 and 2 weeks of storage in dry conditions. The electrode retains 92% and 88% of its initial response, respectively. Such stability seems to be acceptable for most practical applications.
Conclusions
We have shown in this work the advantageous features of carbon paste technology, polymer modification, and dispersion of metallic particles into an organic polymer. A novel electrode has been described herein, consisting of nickel ions loaded into a poly(o-aminophenol)-modified carbon paste electrode by immersion of the polymeric-modified electrode in a nickel ion solution. The Ni/POAP/MCPE can catalyze the oxidation of methanol. The kinetic process of the catalytic reaction can be explained using cyclic voltammetry and chronoamperometry. The values for the rate constant k obtained from the chronoamperometric method indicated that the modified electrode can overcome the kinetic limitations for methanol oxidation by a catalytic process and can decrease the overpotential for the oxidation reaction of methanol.
References
Appleby AJ, Foulkess FR (1989) Fuel cell handbook. Van Nostrand Reinhold, New York Ch 11
Wasmus S, Kuver A (1999) J Electroanal Chem 461:14 doi:10.1016/S0022-0728(98)00197-1
Ren X, Zelenay P, Thomas S, Davey J, Gottesfeld S (2000) J Power Sources 86:111 doi:10.1016/S0378-7753(99)00407-3
Wei Z, Guo H, Tang Z (1996) J Power Sources 58:239 doi:10.1016/S0378-7753(96)02389-0
Lamy C, Lima A, Rhun VL, Coutanceau C, Leger JM (2002) J Power Sources 105:283 doi:10.1016/S0378-7753(01)00954-5
Abdel Rahim MA, Abdel Hameed RM, Khalil MW (2004) J Power Sources 134:60
Verma A, Basu S (2005) J Power Sources 145:282 doi:10.1016/j.jpowsour.2004.11.071
Heli H, Jafarian M, Mahjani MG, Gobal F (2004) Electrochim Acta 49:4999 doi:10.1016/j.electacta.2004.06.015
Jafarian M, Mahjani MG, Heli H, Gobal F, Khajehsharifi H, Hamedi MH (2003) Electrochim Acta 48:423 doi:10.1016/S0013-4686(03)00399-2
Parsons R, Vander Noot T (1988) J Electroanal Chem 257:9 doi:10.1016/0022-0728(88)87028-1
Nishimura K, Machida K, Enyo M (1988) J Electroanal Chem 251:117 doi:10.1016/0022-0728(88)80389-9
Emilia M, Fracisco JC, Joseluis VZ, Antomo A (1992) Electrochim Acta 37:1883 doi:10.1016/0013-4686(92)85094-2
Biswas PC, Nodasaka Y, Enyo M (1996) J Appl Electrochem 26:30 doi:10.1007/BF00248185
Tripkovic AV, Marinkovic N, Popovic KD, Adzic RR (1995) Russ J Electrochem 31:993
Xu CW, Shen PK (2004) Chem Commun (Camb) 19:2238 doi:10.1039/b408589b
Wei ZD, Chan SH (2004) J Electroanal Chem 569:23 doi:10.1016/j.jelechem.2004.01.034
Guo JW, Zhao TS, Prabhuram J, Chen R, Wong CW (2006) J Power Sources 156:345 doi:10.1016/j.jpowsour.2005.05.093
Xu CW, Shen PK (2005) J Power Sources 142:27 doi:10.1016/j.jpowsour.2004.10.017
Hu CC, Liu KY (1999) Electrochim Acta 44:2727 doi:10.1016/S0013-4686(98)00400-9
Tripkovic AV, Popovic KD, Momcilovic JD, Drazic DM (1998) Electrochim Acta 44:1135 doi:10.1016/S0013-4686(98)00216-3
Schell M (1998) J Electroanal Chem 457:221 doi:10.1016/S0022-0728(98)00315-5
Jiang L, Sun G, Sun S, Liu J, Tang S, Li H et al (2005) Electrochim Acta 50:5384
Green CL, Kucernak A (2002) J Phys Chem 106B:106
Safarian HM, Srininvasan R, Chu D, Gilman S (1998) Electrochim Acta 44:1447 doi:10.1016/S0013-4686(98)00268-0
Lio L, Pu C, Viswanathan R, Fan Q, Liu R, Smotkin ES (1998) Electrochim Acta 43:3657 doi:10.1016/S0013-4686(98)00123-6
Ciszewski A, Milczarek G, Lewandowska B, Krutowski K (2003) Electroanalysis 15:518 doi:10.1002/elan.200390062
Agboola B, Nyokong T (2007) Electrochim Acta 52:5039 doi:10.1016/j.electacta.2007.02.017
Samant PV, Fernandes JB (1999) J Power Sources 79:114 doi:10.1016/S0378-7753(99)00043-9
Ureta-Zanartu MS, Alarcon A, Munoz G, Gutierrez C (2007) Electrochim Acta 52:7857 doi:10.1016/j.electacta.2007.06.055
Obirai J, Bedioui F, Nyokong T (2005) J Electroanal Chem 576:323 doi:10.1016/j.jelechem.2004.11.006
Xu C, Hu Y, Rong J, Jiang SP, Liu Y (2007) Electrochem Commun 9:2009 doi:10.1016/j.elecom.2007.05.028
Kulesza PJ, Matczak M, Wolkiewicz A, Grzybowska B, Galkowski M, Malik MA et al (1999) Electrochim Acta 44:2131 doi:10.1016/S0013-4686(98)00321-1
Castro Luna AM (2000) J Appl Electrochem 30:1137 doi:10.1023/A:1004050922065
Mikhaylova AA, Khazova OA, Bagotzky VS (2000) J Electroanal Chem 480:225 doi:10.1016/S0022-0728(99)00464-7
Delime F, Leger JM, Lamy C (1999) J Appl Electrochem 29:1249 doi:10.1023/A:1003788400636
Venancio EC, Napporn WT, Motheo AJ (2002) Electrochim Acta 47:1495 doi:10.1016/S0013-4686(01)00877-5
Golabi SM, Nozad A (2004) Electroanalysis 16:199 doi:10.1002/elan.200302768
Liu SJ (2004) Electrochim Acta 49:3235 doi:10.1016/j.electacta.2004.02.038
Perez-Morales M, Munoz E, Martın-Romero MT, Camacho L (2005) Langmuir 21:5468 doi:10.1021/la0470683
Maximovitch S, Bronoel G (1981) Electrochim Acta 26:1331 doi:10.1016/0013-4686(81)85118-3
Ojani R, Raoof JB, Afagh PS (2004) J Electroanal Chem 571:1 doi:10.1016/j.jelechem.2004.03.032
Ojani R, Raoof JB, Hoseini SR (2007) Electrochim Acta 53:2402
Goncalves D, Faria RC, Yonashiro M, Bulhoes LOS (2000) J Electroanal Chem 487:90 doi:10.1016/S0022-0728(00)00151-0
Hernandez N, Ortega JM, Choy M, rtiz R (2001) J Electroanal Chem 515:123 doi:10.1016/S0022-0728(01)00619-2
Eramo FD, Marioli JM, Arevalo AA, Sereno LE (1999) Electroanalysis 11:481 doi:10.1002/(SICI)1521-4109(199906)11:7<481::AID-ELAN481>3.0.CO;2-7
Casella IG, Cataldi TRI, Guerrieri A, Desimoni E (1996) Anal Chim Acta 335:217 doi:10.1016/S0003-2670(96)00351-0
Pham MT, Maitz MF, Richter E, Reuther H, Prokert F, Mucklich A (2004) J Electroanal Chem 572:185 doi:10.1016/j.jelechem.2004.06.011
Majdi S, Jabbari A, Helli H, Moosavi-Movahedi AA (2007) Electrochim Acta 52:4622 doi:10.1016/j.electacta.2007.01.022
Hajjizadeh M, Jabbari A, Heli H, Moosavi-Movahedi AA, Shafiee A, Karimian K (2008) Anal Biochem 373:337 doi:10.1016/j.ab.2007.10.030
Yi Q, Zhang J, Huang W, Liu X (2007) Catal Commun 8:1017 doi:10.1016/j.catcom.2006.10.009
Hu CC, Cheng CY (2002) J Power Sources 111:137 doi:10.1016/S0378-7753(02)00296-3
Hahn F, Eden BB, Croissant MJ, Lamy C (1986) Electrochim Acta 31:335 doi:10.1016/0013-4686(86)80087-1
Subbaiah T, Mallick SC, Mishra KG, Sanjay K, Das RP (2002) J Power Sources 112:562 doi:10.1016/S0378-7753(02)00470-6
Yevidal AD, Figlarz M (1987) J Appl Electrochem 17:589 doi:10.1007/BF01084134
Jafarian M, Mahjani MG, Heli H, Gobal F, Heydarpoor M (2003) Electrochem Commun 5:184 doi:10.1016/S1388-2481(03)00017-1
Liu B, Zhang Y, Yuan H, Yang H, Yang E (2000) Int J Hydrogen Energy 25:333 doi:10.1016/S0360-3199(99)00026-9
Bard AJ, Faulkner LR (2001) Electrochemical methods. Wiley, New York
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ojani, R., Raoof, JB. & Fathi, S. Nickel–poly(o-aminophenol)-modified carbon paste electrode; an electrocatalyst for methanol oxidation. J Solid State Electrochem 13, 927–934 (2009). https://doi.org/10.1007/s10008-008-0626-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-008-0626-z