
Abstract
A series of energetic compounds were derived from [1,2,4]triazolo[1,5-a][1,3,5]triazine and azo-bridged fused backbone by introducing the –NO2, –NHNO2, –ONO2, –N3, and –NH2 explosophoric groups. The influence of explosophoric groups on energetic properties has been explored. All the compounds exhibit positive energy content (34.4–1955.4 kJ/mol) and densities (1.71–1.99 g/cm3) subject to fused triazole and triazine framework and various functional groups. The designed compounds with –NHNO2, –ONO2, and –NO2 functional groups possess high detonation velocities (8.23–9.00 km/s), pressures (30.94–37.68 GPa), Gurney velocities (2.70–2.88 km/s), and power index (109–131%) superior to TNT (6.94 m/s, 22.0 GPa, 2.37 km/s, and 118%) and comparable with RDX (8.60 km/s, 33.92 GPa, 2.93 km/s, and 169%) and HMX (8.90 km/s, 38.39 GPa, 2.97 km/s, and 169%). Based on high nitrogen and energy content, performance parameters, and sensitivity data, the designed compounds show high potential to be used as energetic materials.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrogen-rich compounds with suitable explosophoric groups gained significant interest because their performance properties can be changed by careful choice of functional groups [1,2,3,4,5,6]. Nitrogenous 5- and 6-membered heterocycles, such as triazole, tetrazole, 1,3,5-triazine, and 1,2,4,5-tetrazine, serve as superior backbones for the search of novel energetic compounds. Among them, the combination of 5- and 6-membered fused rings provides the enormous C–N, N–N, and N=N bonds in the molecular backbone and contributing to high-energy storage and density. The lower carbon and hydrogen percentage in the molecule reduces the need for oxygen during explosion or propulsion reactions and produces nitrogen gas as a major product. As a result, potential fused heterocycles based on a combination of 5- and 6-membered nitrogen-rich rings are reported in the literature [3] (see Fig. 1). Additionally, the incorporation of –NO2, –NHNO2, –ONO2, –N3, and –NH2 functional groups modifies the energetic properties of the entire molecule. Fused heterocyclic rings with coplanar conjugated structure are expected to reconcile the high-performance stability conflict and may provide the energy–safety balanced backbone.

Reported fused heterocyclic backbone
Herein, we report the design of energetic compounds based on [1,2,4]triazolo[1,5-a][1,3,5]triazine (triazolo-triazine) fused backbone by introducing the three functional groups into the molecular scaffold (see Fig. 2). Although a wide variety of energetic materials are designed, the availability of easy and straightforward synthesis route of triazolo-triazine fused backbone [7,8,9] makes the study more feasible and relevant (see Scheme S1). In [5, 6]-bicyclic fused rings, the molecular framework helps to improve nitrogen content and also furnishes positions to add functional groups. The azo (–N=N–) linkage [10,11,12] is incorporated into two triazolo-triazine rings for improving the conjugation and understanding the impact of the molecular backbone on performance. The addition of an amino group is known to diminish the sensitivity, while the azido group contributes to high positive heats of formation. In contrast, nitramine, nitrate-ester, and nitro groups boost oxygen balance, density, and performance [13, 14]. Structural diversification of substituted triazolo-triazine fused backbone has a dramatic effect on their energetic properties, making them potential candidates as energetic green materials.

Molecular structures of designed triazolo-triazine and azo-bridged derivatives
Results and discussion
In this study, the Gaussian 09 program package [15] was used to carry out all computations. The geometries of the triazolo-triazine derivatives were optimised without any symmetry restriction at the B3PW91/6-31G(d,p) level and verified to true local energy minima on the potential energy surface without imaginary frequencies. Reliable theoretical methods and equations were adopted for the estimation energetic properties of triazolo-triazine derivatives that are similar to our earlier studies [16,17,18] and are presented in the Supporting Information.
Heat of formation
The heat of formation (HOF) is commonly considered as an indication of stored energy in an energetic material. The molecular total energies and HOF data for compounds TT1–12 are listed in Table 1. The gas-phase HOFs (HOFGas) of triazolo-triazine derivatives were calculated using isodesmic reaction approach (see Fig. 3). The experimental HOF of triazolo-triazine is unavailable; thus, it was calculated using the G4 method [19, 20] with the atomization reaction approach. The HOF of TT1 fused ring is calculated as 306.7 kJ/mol, found relatively higher than triazole (192.7 kJ/mol) and triazine (225.8 kJ/mol). The addition of –N=N– linkage between triazolo-triazine rings considerably improves the HOF (866.4 kJ/mol). It is observed that all triazolo-triazine compounds retain high positive HOFs, which is an important energetic parameter of high-energy materials. When the substituted functional groups are –NH2, –ONO2, or –NO2, HOF values decrease compared with TT1 and TT7 unsubstituted derivatives. The –NH2,–ONO2, or –NO2 derivatives reduce the HOF of TT1 by 159–172 kJ/mol, while in azo-substituted derivative by 17–472 kJ/mol. For the substituent –NHNO2, the increase or decrease in HOF is not apparent. It is observed that the –N3 group has a significant contribution in raising the HOFs of the triazolo-triazine and its azo unsubstituted derivative boosts the HOF by 887 and 1089 kJ/mol, respectively. The predicted HOFSolid of TT3 and TT9 is 1193.6 and 1955.5 kJ/mol. The influence of explosophoric groups on the energy content has the order of –N3 > –NHNO2 > –H > –NO2 > –NH2 > –ONO2. Overall, high nitrogen content in fused heterocycle and selection of the explosophoric group govern the HOF.

Isodesmic reactions used in the prediction of HOFGas for triazolo-triazine derivatives
Oxygen balance, nitrogen content, and density
Oxygen balance (OB) is an important indication of the availability of oxygen in the molecular framework and degree to which a molecule can be oxidised. Better/positive OB value ensures the higher gaseous detonation products and results in a greater energetic performance. In CHNO energetic molecules, positive or close to zero OB values are desirable to reduce the formation of toxic carbon monoxide. It is found that, except TT5, all the derivatives show negative OB, i.e. oxygen-deficient nature. The OB values for triazolo-triazine derivatives range from − 125 to 5%, while for its azo-substituted derivatives fall in the range of − 12 to − 107% (see Table 2). The oxygen-containing explosophoric groups (–NHNO2, –ONO2, and –NO2) are more helpful in achieving better OB values. All the designed compounds possess moderate to high nitrogen content. The nitrogen content in triazolo-triazine derivatives ranged from 36 to 80%, wherein azo-substituted derivatives show 43 to 78% (see Table 2). The –N3 substituted compounds show the highest nitrogen content compared with other derivatives.
Density is one of the major parameters in explosives deciding its energetic performance. Denser materials are also essential to pack a maximum amount of material in limited space. The computed densities of the triazolo-triazine derivatives ranged from 1.57 to 1.95 g/cm3, whereas azo derivatives vary from 1.71 to 1.99 g/cm3 (see Table 2). Compounds TT5, TT6, TT10, and TT11 possess a density above 1.90 g/cm3. Among 12 designed compounds, eight molecules reveal greater densities than RDX (1.80 g/cm3), and four molecules have densities superior to HMX (1.90 g/cm3). Compared with the unsubstituted molecular backbone (TT1 and TT7), introducing various functional groups helps to gain better density. Our results show that the –NHNO2, –ONO2, and –NO2 groups contribute to density to a greater extent over –NH2 and –N3 groups.
Performance parameters
The detonation velocity (D) and pressure (P) of the triazolo-triazine derivatives were computed using Kamlet-Jacobs equations [21] and are listed in Table 2. The predicted D and P values of the triazolo-triazines fall in the range 5.26–9.00 km/s and 11.88–37.68 GPa, respectively, whereas the azo-linked derivatives show D and P values of 6.12–8.72 km/s and 16.62–35.72 GPa, respectively. Molecules with –ONO2 functional group (TT5 and TT11) show the highest detonation performance among the designed compounds due to their high densities and OB. The high-density and good OB also help to achieve admirable detonation parameters in TT4 (D = 8.54 km/s, P = 33.18 GPa), TT5 (D = 9.00 km/s, P = 37.68 GPa), and TT10 (D = 8.72 km/s, P = 35.72 GPa), which are comparable or superior to RDX (D = 8.60 km/s, P = 33.9 GPa). This shows that the –NHNO2, –ONO2, and –NO2 groups are competent explosophoric unit for achieving better detonation performance and marked the advantage of oxygen-rich groups in energetic molecules.
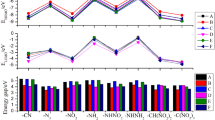
The explosive power index (PI) mainly depends on the volume of gases produced during detonation and the heat of detonation and relative to picric acid [22]. The calculated PI values for triazolo-triazine derivatives vary from 21 to 131% (see Table 2). The decomposition products are assumed via the Kistiakowsky-Wilson rules [22] and are presented in Table S4. TT3–6 and TT11 are more potent than TNT (116%). Most of the triazolo-triazine derivatives show poor explosive power index due to their negative oxygen balance, which leads to forming fewer amounts of gaseous CO, CO2, and H2O. Gurney velocity (\( \sqrt{2E} \)) is a valuable property to represent the ballistic performance and energy outcome from energetic material. Gurney velocities are predicted using Kamlet-Finger formula [23] and validated with Hardesty-Kennedy approach [24] based on their molecular mass, density, heat of detonation, and gaseous combustion products per gram of explosive. Except –H and –NH2 substituted molecules, all the triazolo-triazine derivatives have \( \sqrt{2E} \) values higher than TNT (2.37 km/s) owing to their HOFs, high nitrogen content, better oxygen balance, and densities (see Table 2). The triazolo-triazine derivatives with –NHNO2, –ONO2, and –NO2 groups show \( \sqrt{2E} \) values ranged from 2.70 to 2.92 km/s, which are comparable to or higher than RDX (2.83 km/s). Figure 4 compares the calculated densities D, P, and \( \sqrt{2E} \) for the triazolo-triazine derivatives together with TNT and RDX. Due to high detonation performance, the triazolo-triazine derivatives with –NHNO2, –ONO2, and –NO2 groups may be considered as the candidate molecules carrying the maximum potential for synthesis and usage in high-density energetic materials. The relationship among the contributions of various explosophoric groups to the performance parameters is –ONO2 > –NHNO2 > –NO2 > –N3 > –H > –NH2.

Comparison of densities and performance parameters of triazolo-triazine derivatives with TNT and RDX
Sensitivity trend
Reliable sensitivity assessment of newly designed molecules with existing benchmark explosives has great importance because safe handling, storage, and safety aspects are of utmost importance in practical use. The sensitivity of explosives to heat, impact, friction, or other external stimuli depends on the molecular backbone, functional groups, physical nature, environmental conditions, etc.; hence, it is challenging to employ particular and uniform correlations. To assess the sensitivity of the triazolo-triazine derivatives, we examined the free space in a unit cell per molecule (ΔV), energy gap (ΔE) between the highest occupied and lowest unoccupied molecular orbitals, and heat of detonation (Q) and compared them with benchmark explosives RDX and HMX. Politzer et al. [25,26,27,28,29] suggested that higher ΔV value corresponds to a more sensitive compound. Calculated ΔV, ΔE, and related parameters are listed in Table 3. Except for TT1 (31.4 Å3), TT2 (29.3 Å3), and TT8 (38.9 Å3), all other derivatives show higher ΔV values than RDX (45.6 Å3) and HMX (49.2 Å3) and are expected to be sensitive. Among the designed compounds, the azo-bonded compounds (TT9–12) show ΔV value higher than 90 Å3; this may be attributed to more explosophoric functional groups present in the molecular framework.
In the literature [30,31,32], the energy gap (ΔE) is linked with chemical reactivity, stability, and sensitivity of the energetic materials, where higher ΔE value indicates lower sensitivity of the molecule. It is clear from the ΔE values that incorporation of –NHNO2, –ONO2, –NO2, and –N3 functional groups decreases the energy gap and is responsible for the higher sensitivity of the molecules. The unsubstituted molecules (TT1, TT7) and molecule with –NH2 groups (TT2, TT8) show better insensitivity. It is also observed that all the azo-linked compounds are more sensitive than triazolo-triazine derivatives. Overall, all the triazolo-triazine derivatives have lower ΔE values compared with RDX (6.43 eV) and HMX (6.12 eV) and are expected to be sensitive.
Politzer et al. [33] reported the possible link between sensitivity of explosives and the Q value, where higher Q value corresponds to greater sensitivity. Table 3 lists the Q values calculated using the Kamlet-Jacobs formula [21]. It is observed that Q values show a slightly different trend in sensitivity compared with ΔV and ΔE values. According to the Q values, all the designed triazolo-triazine derivatives are anticipated to be insensitive than RDX (1500 cal/g). The calculated Q values for triazolo-triazine derivatives range from 341 to 1418 cal/g. Overall, the analysis of ΔV, ΔE, and Q values indicates that incorporation of –NH2 groups and reducing the trigger linkages (C–N3, –NH–NO2, C–ONO2, and C–NO2) will help to achieve better insensitivity in the target molecules.
Conclusions
We have designed a new series of energetic materials comprising triazolo-triazine fused backbone by introducing the –NH2, –NHNO2, –ONO2, –NO2, and –N3 groups. All designed compounds reveal high energy content, consistent detonation performance, and stability. Compounds TT3–7 and TT9–12 display good densities (1.88–1.99 g/cm3), high detonation velocities (8.23–9.00 km/s), and pressures (30.94–37.68 GPa), which are superior to TNT and equivalent to RDX. We conclude that the triazolo-triazine fused backbone will improve the energy content of the entire molecule and, when paired with –NHNO2, –ONO2, and –NO2 groups, achieve high detonation performance. The –NH2 and –N3 groups are recognised to be good moieties for improving stability and energy content, respectively (see Fig. 5). Based on the high detonation properties, good densities, and energy content, triazolo-triazine derivatives are identified as potential energetic compounds.

Effect of various explosophoric groups on energy content, performance, and stability of triazolo-triazine derivatives
References
Gao H, Shreeve JM (2011) Azole-based energetic salts. Chem Rev 111:7377–7436
Klapötke TM (2012) Chemistry of high-energy materials2nd edn. De Gruyter, Berlin
Gao H, Zhang Q, Shreeve JM (2020) Fused heterocycle-based energetic materials (2012–2019). J Mater Chem A 8:4193–4216
Ma J, Tang Y, Cheng G, Imler GH, Parrish DA, Shreeve JM (2020) Energetic derivatives of 8-nitropyrazolo[1,5-a][1,3,5]triazine-2,4,7-triamine: achieving balanced explosives by fusing pyrazole with triazine. Org Lett 22:1321–1325
Chen S, Liu Y, Feng Y, Yang X, Zhang Q (2020) 5,6-Fused bicyclic tetrazolo-pyridazine energetic materials. Chem Commun 56:1493–1496
Politzer P, Murray JS (2017) Computational analysis of polyazoles and their N-oxides. Struct Chem 28:1045–1063
Berecz G, Pongó L, Kövesdi I, Reiter J (2002) On triazoles XLV [1] synthesis of 5,7-diamino-1,2,4-triazolo[1,5-a] [1,3,5]triazines. J Heterocyclic Chem 39:327–334
Dolzhenko AV, Dolzhenko AV, Chui WK (2007) Synthesis of 5,7-diamino[1,2,4]triazolo[1,2-a][1,3,5]triazines via annulation of 1,3,5-triazine ring onto 3(5)-amino-1,2,4-triazoles. Heterocycles 71:429–436
Ma J, Cheng G, Ju X, Yi Z, Zhu S, Zhang Z, Yang H (2018) Amino-nitramino functionalized triazolotriazines: a good balance between high energy and low sensitivity. Dalton Trans 47:14483–14490
Hu L, Yin P, Zhao G, He C, Imler GH, Parrish DA, Gao H, Shreeve JM (2018) Conjugated energetic salts based on fused rings: insensitive and highly dense materials. J Am Chem Soc 140:15001–15007
Tang Y, Yang H, Shen J, Wu B, Ju X, Lu C, Cheng G (2012) Synthesis and characterization of 1,1′-azobis(5-methyltetrazole). New J Chem 36:2447–2450
Wang F, Du H, Zhang J, Gong X (2011) Comparative theoretical studies of energetic azo s-triazines. J Phys Chem A 115:11852–11860
Liu H, Wang F, Wang GX, Gong XD (2012) Theoretical investigations on structure, density, detonation properties, and sensitivity of the derivatives of PYX. J Comput Chem 33:1790–1796
Wei T, Zhu W, Zhang X, Li Y, Xiao H (2009) Molecular design of 1,2,4,5-tetrazine-based high-energy density materials. J Phys Chem A 113:9404–9412
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Kurant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salwador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09, Revision E.01. Gaussian Inc., Wallingford
Ghule VD (2012) Computational studies on energetic properties of trinitro-substituted imidazole−triazole and pyrazole−triazole derivatives. J Phys Chem A 116:9391–9397
Ghule VD (2013) Studies on energetic properties for nitrotetrazole substituted triazole and oxadiazole derivatives with density functional theory. Mol Phys 111:95–100
Nirwan A, Devi A, Ghule VD (2019) Theoretical determination of the effects of various linkages between trinitrobenzenes on energetic properties and sensitivity. J Mol Model 25:315
Curtiss LA, Redfern PC, Raghavachari K (2007) Gaussian-4 theory. J Chem Phys 126:084108
Rayne S, Forest K (2010) Estimated gas-phase standard state enthalpies of formation for organic compounds using the Gaussian-4 (G4) and W1BD theoretical methods. J Chem Eng Data 55:5359–5364
Kamlet MJ, Jacobs SJ (1968) Chemistry of detonations. I. A simple method for calculating detonation properties of C−H−N−O explosives. J Chem Phys 48:23–36
Akhavan J (2004) The chemistry of explosives2nd edn. RSC Paperbacks, Cambridge
Kamlet MJ, Finger M (1979) An alternative method for calculating gurney velocities. Combust Flame 34:213–214
Hardesty DR, Kennedy JE (1977) Thermochemical estimation of explosive energy output. Combust Flame 28:45–59
Politzer P, Murray JS (2014) Impact sensitivity and crystal lattice compressibility/free space. J Mol Model 20:2223
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2011) Sensitivity and the available free space per molecule in the unit cell. J Mol Model 17:2569–2574
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2010) A possible crystal volume factor in the impact sensitivities of some energetic compounds. J Mol Model 16:895–901
Politzer P, Murray JS (2016) High performance, low sensitivity: conflicting or compatible? Propellants Explos Pyrotech 41:414–425
Politzer P, Murray JS (2015) Some molecular/crystalline factors that affect the sensitivities of energetic materials: molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J Mol Model 21:25
Morrill JA, Byrd EFC (2008) Development of quantitative structure–property relationships for predictive modeling and design of energetic materials. J Mol Graph Model 27:349–355
Wang W, Zhu W, Li J, Cheng B, Xiao H (2013) Periodic density functional theory study of the high-pressure behavior of energetic crystalline 1,4-dinitrofurazano[3, 4-b]piperazine. J Mol Model 19:305–314
Khan RU, Zhu S, Zhu W (2019) DFT studies on nitrogen-rich pyrazino [2, 3-e] [1, 2, 3, 4] tetrazine–based high–energy density compounds. J Mol Model 25:283
Politzer P, Murray JS (2015) Impact sensitivity and the maximum heat of detonation. J Mol Model 21:262
Acknowledgements
Anjali thanks UGC-CSIR, Ministry of Human Resource Development, Government of India for Junior Research Fellowship. RSM thank Armament Research Board, Defence R&D Organization, DRDO for research fellowship (No. ARMREB/CDSW/2019/211).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Computational details, selective structural parameters, and energetic properties related parameters of triazolo-triazine derivatives are given in the Supporting Information. (DOCX 1475 kb)
Rights and permissions
About this article

Cite this article
Maan, A., Mathpati, R.S. & Ghule, V.D. Substituted triazolo-triazine derivatives as energetic materials: a computational investigation and assessment. J Mol Model 26, 184 (2020). https://doi.org/10.1007/s00894-020-04455-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04455-9