
Abstract
Taxanes (paclitaxel, docetaxel, cabazitaxel) are anticancer drugs as microtubule inhibitors. Following our previous studies on paclitaxel and docetaxel, in this work, we examine cabazitaxel and compare these three taxenes. The binding interaction of three taxanes with various β-tubulin isotypes is studied by homology modeling, molecular docking, and molecular dynamics simulations. The results show that the effects of docetaxel on βI-tubulin (− 29.5 kcal/mol) and of paclitaxel on βIIa-tubulin (− 25.5 kcal/mol) are much stronger than their effects on βIII-tubulin (− 17.8 kcal/mol and − 8.6 kcal/mol, respectively). However, the effect of cabazitaxel on βIII-tubulin (− 23.0 kcal/mol) is comparable with that on βI-tubulin (− 24.0 kcal/mol) and βIIa-tubulin (− 25.9 kcal/mol), consistent with the fact that overexpression of βIII-tubulin increases the drug resistance to paclitaxel and docetaxel, but has little influence for cabazitaxel. This theoretical research supports the use of cabazitaxel for patients who are resistant to the action of paclitaxel and docetaxel.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
According to the 2018 National Cancer Report, the highest morbidity among men is prostate cancer, and that among women is breast cancer considering all malignancies in the US, and they rank second only to heart disease [1]. The latest cancer data show that morbidity and mortality of various malignant tumors are rising, and tumors are likely to remain one of the major causes of death [2,3,4,5].
Microtubules (MTs), made up of polymerized α- and β-tubulin dimers, play a very important role in many cellular processes, including the cancer cellular process [6]. MTs are involved in the development and maintenance of cell shape, cell motility, intracellular transport, cell division, and mitosis [7, 8]. Microtubule inhibitors (MTIs) such as taxanes, epothilones, vinca alkaloids, and colchicines effectively block cancer cell division by changing the microtubule dynamics (either assembly or disassembly) required for proper mitosis. The action sites of the classical MTIs are shown in Fig. 1 [8,9,10,11,12,13,14].

Taxane anticancer drugs, a class of diterpenes, are common drugs of MTIs [17]. There are mainly three analogs used in clinical applications, namely, paclitaxel (PTX), docetaxel (DTX), and cabazitaxel (CTX). The binding sites of taxanes are on β-tubulin as shown in Fig. 1 [18,19,20,21,22,23,24]. PTX is the first generation of taxanes. In 1992, it was approved for the treatment of advanced non-small cell lung cancer, breast cancer, and ovarian cancer by the Food and Drug Administration (FDA) [17, 25], and recently, it has also been used to treat Kaposi sarcoma, cervical cancer, and pancreatic cancer [26,27,28]. Subsequently, researchers synthesized a second-generation compound DTX, with better water solubility and lesser adverse reactions, by modifying the PTX side chains [29]. In 1996, DTX was approved by the FDA to treat early or advanced breast cancer [30], and it is the main chemotherapy drug used in current clinical treatments for the metastatic castrate-resistant prostate cancer (mCRPC) [31]. DTX is found to be effective for most patients, but it is easy to develop tolerance [32]. To solve the drug resistance problem, researchers synthesized a third-generation compound CTX, which was approved by the FDA in 2010 to treat the metastatic prostate cancer [33, 34], and CTX is used as a second-line treatment for mCRPC patients [35, 36].
There are many tubulin isotypes (such as βI-tubulin, βII-tubulin, and βIII-tubulin) in the human body, and the identified β-tubulin isotypes are expressed differently in different cell types [37,38,39,40]. In particular, the expression of βIII-tubulin isotype is often associated with drug-resistant and aggressive cancers [41,42,43,44]. It was reported that the amount of βIII-tubulin was relatively low at the early time of treatment, and PTX and DTX were effective [45,46,47]. However, after the treatment, the amount of βIII-tubulin increases, and then, the effects of PTX and DTX begin to wear off [32, 48]. Smiyun et al. reported that CTX was effective for those patients who were resistant to DTX [49].
Since there are still many limitations in clinical tests [50,51,52,53,54,55], the detailed action mechanism of different isotypes with taxanes remains unclear. In the present study, we will theoretically explore drug-binding modes and drug resistance of taxanes interacting with different tubulin isotypes. Our previous studies [24, 56] have predicted the lowest-lying conformations of the PTX and DTX with Density Functional Theory (DFT) methods. In the present paper, we will optimize the structure of CTX with the same methods. We will further build three-dimensional models of the human tubulin isotypes by homology modeling and then examine the tubulin isotypes with higher binding affinities to taxanes using the molecular docking method. Since the drug binding on the protein and the molecular recognition is a dynamic process [57], we will also investigate the detailed active sites and binding affinities of these processes using molecular dynamics (MD) simulations.
Computational methods
Optimization molecular structure of CTX
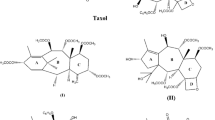
The taxanes studied in the present paper include PTX, DTX and CTX (Fig. 2). The optimized geometries of PTX and DTX have been reported previously [24, 56], and the molecular geometry of CTX was optimized here. The initial structure of CTX was generated by modifying the lowest-lying DTX structure. The Gaussian16 software [58] was used to search the possible global minima. To avoid missing any promising structures, the full conformational space of gaseous CTX was explored through a systematic variation of rotational degrees of freedom around seven bonds in the C13 side chain, as shown in Fig. 3. The dihedral angle for each bond can be varied from 0 to 360° with the increment of 60° for six asymmetric rotations or the increment 120° for one symmetrical rotation. Thus, 139,968 initial conformations were generated.

Structures of PTX (A), DTX (B), and CTX (C). The parental ring structures of three taxanes are the same, but the C7, C10, and C13 side chains are different

The degrees of freedom for CTX. For the C13 side chain of CTX, a total of seven rotatable bonds (a–g) were selected. We chose 60° as the incremental rotation for each rotational torsion angle except for (e); the increment of which is 120° due to the local symmetry
We initially used the semi-empirical PM7 method, and then, the HF/3-21G* method to remove distracting conformations such as those with imaginary vibrational frequencies. Then, we used the B3LYP/6-311G* method to obtain gaseous conformations. The vibrational frequency analysis was carried out at the same level of theory. More accurate relative energies were obtained from single-point energies at the M06-L/6-311G(2df,p) level in both the gaseous phase and water solution (with the polarizable continuum model). Finally, the excited states of the lowest-lying CTX conformation were studied with the TD DFT method, namely, the CAM-B3LYP/6-311+G(2df,p).
Homology modeling
Homology modeling is a method to construct an atomic-resolution model of the “target” protein from its amino acid sequence and an experimentally known three-dimensional structure of a related homologous “template” protein [59]. It can produce high-quality structural models when the target and template are closely related.
In the present study, we adopted homology modeling to build the three-dimensional models for different isotypes of the human αβ-tubulin. The “template” αβ-tubulin, including GTP (Guanosine triphosphate), GDP (Guanosine iphosphate), PTX, and Mg2+ bound to it, was taken from the high-resolution cryo-electron microscopy structure of microtubule (PDB ID: 5SYF.pdb, resolved at 3.5 Å, shown in Fig. S1 in Supplementary Material) [22]. The amino acid sequences of the human tubulin were collected from the UniProt protein sequence database (www.uniprot.org) [60]. We used the online tool Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo) for multiple sequence alignment analysis [61] and then used SWISS-MODEL (www.swissmodel.expasy.org) for single template modeling [62,63,64,65,66].
Molecular docking
Molecular docking is a method of molecular simulation to predict the preferential orientation of one molecule binding with another when they form a stable complex. It is one of the most common methods to predict the binding conformation of a ligand with a suitable protein target [67].
In the present study, molecular docking was performed using the AutoDock4.2 software [68]. At first, a larger box with the 126 × 126 × 126 grid and the grid spacing of 1.0 Å was adopted for the global docking of the αβ-tubulin dimer with CTX to find the binding sites of CTX on tubulin dimer. And then, the different human tubulin isotypes with the lowest-lying PTX, DTX and CTX conformations were used for the local docking on the binding sites we found. The local docking was performed with a smaller box with a finer grid of 60 × 60 × 60 and the grid spacing of 0.375 Å. Using the Lamarckian Genetic Algorithm (LGA) for generating binding poses, we set the maximum number of energy evaluations to be 25 million. A total of 100 runs were performed, and for every independent run, a maximum number of 27,000 generations were investigated on a single population of 150 individuals.
Molecular dynamics simulations
The MD simulations were performed for the complexes with high-binding energy between the three taxanes and different tubulin isotypes with the AMBER99SB-LIDN force field from the GROMACS5.0 software [69]. The general Amber force field (GAFF) for PTX, DTX, and CTX was obtained from the antechamber module of AmberTools12 [70]. The systems were immersed in a dodecahedral box of SPC/E waters during simulation [71], where there was a buffer of 1.0 nm between the protein atoms and the edge of the water box. We added Na+ or Cl− ions to make them neutral. The steepest descent method was used for energy minimization. Then NVT ensemble, whose positions were restrained by the LINC algorithm [72], and NPT ensemble, whose temperature was increased from 0 to 300 K under the standard atmospheric pressure, were performed for 1 ns, respectively, and finally, the production simulation was performed for 100 ns with a time step of 2 fs. According to literatures [32, 41, 47, 73,74,75], the drug effect became weak if βIII-tubulin overexpressed in cancer cells. In order to rationalize this phenomenon, the same MD simulations were also performed for the complexes of βIII-tubulin and three taxanes. The details are presented in Table S1 (Supplementary Material).
With the MD results, we could further assess the free binding energies between β-tubulin and taxanes.
where Gcomplex, Gfree-protein, and Gfree-ligand are free energies for the complex, protein, and ligand, respectively. The MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method [76, 77] implemented in the g_mmpbsa program [78] is adopted, and the parameters used here are presented in Table S1 (Supplementary Material). The method of total free energy calculation is referred to [76, 77], which is the summation of gas phase energy and the solvation energy. Note that the solvation free energy can be decomposed into polar and non-polar contributions. For the polar solvation energy, the implicit solvent model (ɛ = 2.0) was used. (The other values of dielectric constant were also tried, but the conclusions are essentially the same.) For the non-polar solvation energy, the solvent-accessible surface area (SASA) model was used.
Results and discussion
Optimization molecular structure of CTX
After the PM7 and HF screening, 100 conformations were left out of the initial 139,968 conformations generated by the internal rotations as shown in Fig. 3. Finally, the 20 most stable CTX conformations in the gas phase were obtained at the B3LYP/6-311G* level. The single-point energies were evaluated at the M06-L/6-311G(2df,p) level in both the gas phase and aqueous solution. Table 1 shows the theoretical relative energies, the zero point vibrational energies (ZPVE), the rotational constants, and the dipole moments for these 20 conformations. The lowest-lying CTX conformation (conformation 1, shown in Fig. 4) was found to be the global minimum in both gas phase and solution, and conformation 1 was used for the subsequent studies.

The lowest-lying CTX conformation (conformation 1) in the gas phase at the B3LYP//6-311G* level. The important hydrogen bonds are represented by dotted lines
The hydrogen bonds in the lowest-lying CTX conformation (conformation 1), taking a cutoff distance of 2.8 Å between the H-bonding donor and acceptor, are shown in Fig. 4. It is found that the main hydrogen bond acceptors in CTX are O and N atoms, and the main donors are –NH and –OH groups. The weak hydrogen bonds with the –CH groups as H-bonding donors are also found. As shown in Fig. 4, there are 17 hydrogen bonds in conformation 1. To discuss these hydrogen bonds, we compared them in conformation 1 with those in other conformations (conformations 2 and 3, Fig. S2 in Supplementary Material). There are two differences among these three conformations. At first, the –NH donor in conformation 1 forms two hydrogen bonds with the O atom of C4′-O-C5′ and C1′ = O (with distances of 2.33 Å and 2.22 Å), while the –NH group in conformations 2 and 3 only interacts with the O atom of C4′-O-C5′ (with distances of 2.32 Å and 2.33 Å, respectively). Secondly, the –OH donor on C2′ forms two conventional hydrogen bonds with the O atom of C13–O–C1′ and C=O (linked to C4) in conformation 1 (with the distances of 2.11 Å and 2.18 Å) and in conformation 3 (with the distances of 2.19 Å and 2.03 Å), while the –OH on C2′ in conformation 2 only interacts with the O atom of C=O (linked to C4) by 2.06 Å. It may be further inferred that, with significant structural flexibility in larger biomolecules, the coexistence of dihydrogen bonds, as well as many red-shifting hydrogen bonds, should be a common phenomenon and have important effects on the conformational energetics and other properties.
Our theoretical IR spectra for the lowest three conformations predicted with the B3LYP/6-311G* method (scaled by a factor of 0.96) [79] are shown in Fig. 5. The values for harmonic vibrational frequencies with strong infrared intensities for several representative CTX conformations are also reported in Table 2. Figure 5a shows that there are a number of strong absorption peaks in the range of 1000 to 1300 cm−1, which are corresponding to the normal modes assigned as the C–O–C stretching, benzene ring deformation, and methyl group wagging. Our theoretical results are basically in agreement with the experimental IR observation of 1270, 1246 cm−1 assigned for the C–O–C (ester) stretching modes and of 1170, and 1026 cm−1 assigned for the C–O–C (ether) stretching modes [80]. The vibrational frequencies ranged from 2850 to 3100 cm−1 (Fig. 5b) are corresponding to the assignments of the C–H stretching in methyl, methylene, and methoxy groups, which agree with the experimental value at 2822 to 2980 cm−1 [80]. These C–H stretching modes in different conformations are almost identical, since the C–H bonds are usually transferable among molecules.

Our predicted IR spectra of the three lowest-lying CTX conformations at the B3LYP/6-311G* level: a in the range of 0–1900 cm−1 and b in the range of 2700–3800 cm−1
However, the frequencies related to the O–H stretching modes have an obvious shift among the different conformations, and this reflects the big change of bond strength when H-bond formation occurs. As shown in Table 2, the stretching frequency of O–H on C2′ in conformation 1 (3559 cm−1) is blue-shifted by about 34 cm−1 relative to that in conformation 3 (3525 cm−1), indicating a stronger hydrogen bond between the O–H and C=O (linked to C4) groups in conformation 3. The O–H stretching frequency on C1 in conformation 1 (3591 cm−1) is red-shifted by about 45 cm−1 relative to that in conformation 3 (3632 cm−1) and conformation 13 (3637 cm−1), due to the hydrogen bond between the O–H and the C=O (in PhCOO–) groups in conformation 1.
Table 2 shows that five strong IR frequencies of C=O stretching modes for conformation 1 are in the range of 1682–1718 cm−1. For other conformations, the ranges of these five frequencies are almost the same, and these values are in agreement with the experimental assignment for CTX, which is a very strong peak at 1723 cm−1 [80]. When a C=O group is involved in a hydrogen bond, this C=O stretching frequency would be red-shifted. For example, the C=O frequency is red-shifted by 36 cm−1 for the C9 = O bond compared with that for the C=O bond linked on C2, because a stronger hydrogen bond is for the former.
The ultraviolet absorption of the molecule reflects the electronic transitions to the higher excited states. We explored the three lowest excited electronic states of the CTX (conformation 1) with the time-dependent (TD) method at the CAM-B3LYP/6-311+G(2df,p) level in methanol solution. Table 3 reports the excited energies, dipole transition oscillator strengths, excitations, and types of orbital transitions of the lowest-lying CTX. The orbitals involved in these excitations are displayed in Fig. S3 (Supplementary Material). Although some of these orbitals are delocalized on the phenyl rings as well as on the N and O atoms, we can approximately assign HOMO-4, HOMO-5 as π orbital on the benzene rings, while HOMO, HOMO-3, HOMO-4, HOMO-5, HOMO-7, HOMO-11, HOMO-12, HOMO-13, HOMO-15, HOMO-16 orbitals have components of the N and O lone pairs (symbol n in Table 3). The virtual orbitals LUMO and LUMO+3 are the π* orbitals on the phenyl ring, and the orbitals LUMO, LUMO+1 have components of the σ* orbital on the C and O atoms. The UV spectrum of the three excitations for CTX is predicted to be 228–291 nm (Table 3). The S0–S1 transition at 4.25 eV (291 nm) is an n → σ* transition with weak oscillator strength (0.0028). The S0–S2 transition at 5.10 eV (243 nm) is a mixed nσ*/πσ*/nπ*/ππ* transition, and the S0–S3 transition at 5.43 eV (228 nm) is also a mixed nσ*/πσ*/nπ*/ππ* transition.
Sequence analysis and homology modeling
Nine human β-tubulin sequences were collected from the UniProt database [60], namely, βI-tubulin isotype (UniProt ID = Q9H4B7), βIIa-tubulin isotype (UniProt ID = Q13885), βIIb-tubulin isotype (UniProt ID = Q9BVA1), βIII-tubulin isotype (UniProt ID = Q13509), βIVa-tubulin isotype (UniProt ID = P04350), βIVb-tubulin isotype (UniProt ID = P68371), βV-tubulin isotype (UniProt ID = P07437), βVI-tubulin isotype (UniProt ID = Q9BUF5), and βVIII-tubulin isotype (UniProt ID = Q3ZCM7), and they all were used as target proteins. With the template protein of αβ-tubulin dimer (PDB ID = 5SYF.pdb [22] UniProt ID = P02554) taken from the cryo-electron microscopy structure, the results of multiple alignments are shown in Table 4 and Table S2 (Supplementary Material). The identities between the target proteins and the template protein are very high, all larger than 90% except for βI-tubulin (78%) and βVIII-tubulin (88%). The identity of βII-tubulin to the template is even as high as 98.7%. It was suggested that high-accuracy comparative models are based on more than 50% sequence identity [81]. Therefore, these target proteins in our work are reliable for homology modeling to the construction of the 3D structure models.
The homology modeling for the nine αβ-tubulin isotypes was done with the online tool SWISS-MODEL [62,63,64,65,66]. The results from the homology modeling were also assessed by the Verify-3D program [82, 83]. The good scores (> 94%, Table 4) of the assessment for the nine human β-tubulin isotypes further indicate that all these structures are reliable for the docking studies.
Molecular docking
The preliminary global docking results for the 20 lowest-lying CTX conformations with the αβ-tubulin dimer are shown in Fig. 6 and Table S3 (Supplementary Material). Eighteen CTX conformations are located on β-tubulin (N1), one (conformation 18) located on αβ-tubulin dimer interface (N2), and one (conformation 5) on α-tubulin (N3). As shown in Table S3, the binding energies of CTX conformations docked on β-tubulin (N1) are stronger than that on αβ-tubulin dimer interface (N2) or on α-tubulin (N3). For example, the binding energy of conformation 1 docked on β-tubulin is − 9.74 kcal/mol, while those on αβ-tubulin dimer interface and on α-tubulin are − 7.68 kcal/mol and − 6.58 kcal/mol, respectively. Similar to the previous research on PTX and DTX [16, 19,20,21,22,23,24], the taxanes, including CTX, are found to act mainly on β-tubulin, so the further local docking is simply carried out between the taxanes and different isotypes of β-tubulin.

The binding sites of the lowest-lying 20 CTX conformations on αβ-tubulin dimer found by docking, where N1 is located on β-tubulin, N2 is located on αβ-tubulin dimer interface, and N3 is located on α-tubulin
The local docking results of PTX, DTX and CTX with different β-tubulin isotypes, including the binding affinities and the residues related to the formation of hydrogen bonds, are shown in Table 5. The most common human β-tubulin isotypes (βI-tubulin and βII-tubulin) have significant binding affinities (Table 5). For example, the three strongest binding affinities for PTX are onto βI-tubulin, βIIa-tubulin, and βIIb-tubulin (− 9.01, − 9.84, and − 9.98 kcal/mol, respectively), those for DTX are onto βI-tubulin, βIIa-tubulin, and βIIb-tubulin (− 8.83, − 8.60, and − 8.85 kcal/mol, respectively), and those for CTX are onto βIIa-tubulin, βIIb-tubulin, and βIII-tubulin (− 8.69, − 8.55, and − 8.52 kcal/mol, respectively). Generally speaking, among these β-tubulin isotypes, βI-tubulin and βII-tubulin have stronger binding affinities with the taxanes, indicating that they are the leading action of proteins with taxanes. Our docking results show that βIII-tubulin isotype has weaker binding energy than βI-tubulin isotype with PTX and DTX, and this confirms the experimental studies that the overexpression of βIII-tubulin may reduce the action of taxanes [32, 41, 47, 73,74,75].
Docking modeling provides the number of hydrogen bonds formed between the taxanes and tubulins (Table 5). For βIIa-tubulin, there are two hydrogen bonds docking with PTX and only one with DTX and CTX. The binding affinity of PTX (− 9.84 kcal/mol) with βIIa-tubulin is accordingly stronger than those of DTX and CTX (− 8.60 and − 8.69 kcal/mol). Table 5 also shows that the main acting sites in all β-tubulin isotypes docking with the three taxanes are the amino acid residues His227, Pro272, Thr274, Arg276, Arg359, and Gly360. More detailed docking information is available in Table S4 (Supplementary Material), including the bond geometry parameters and the bond energies.
According to the docking results, the βI-tubulin and βIIa-tubulin isotypes have stronger binding affinities, and they were selected to perform the following MD simulations.
Molecular dynamics simulations
In order to observe the detailed dynamic process of drug binding, three β-tubulin isotypes (βI-tubulin, βIIa-tubulin, βIII-tubulin plus a bound GDP) with three taxanes (PTX, DTX, CTX) were used to construct nine simulation systems. The initial structures were taken from the docking results with the strongest binding affinity, and the MD simulations were carried out for 100 ns. The root mean square deviation (RMSD) analysis suggests that all systems reached their equilibrium structures (Fig. S4 in Supplementary Material), and the deviations in the range of 0.12–0.22 nm indicate that the simulation results are consistent with docking.
For analysis of the complex structures, we evaluated the root mean square of fluctuations (RMSF) for all systems [84]. The RMSF values describe the deviation of an atom in the β-tubulin (Cα atom in the present study) between its instant position and the reference position, which are averaged over the time of simulations. The RMSF values for βI-tubulin, βIIa-tubulin, and βIII-tubulin are shown in Fig. S5 (Supplementary Material). The larger RMSF values indicate highly flexible regions, while the smaller RMSF values indicate constrained regions. For the same β-tubulin isotype, the fluctuation patterns are almost the same despite different taxanes, indicating that the action processes for PTX, DTX and CTX are similar.
Most of RMSF peaks are corresponding to the amino acid residues in the random coil structures, which are closed to the ligands and the GDP molecule. As shown in Fig. S5, the amino acid segments 276–284 in all three taxanes (βI-tubulin, βIIa-tubulin, and βIII-tubulin) have large RMSF values. However, the RMSF values for the 173–178 amino acid segments in βIII-tubulin are higher than those in βIIa-tubulin. For the 37–42 amino acid segments, there are high RMSF values for βI-tubulin, but low values for βIII-tubulin. This shows that the ligand has different interactions with the residues on those segments in βIII-tubulin.
Figure S6 (Supplementary Material) shows the number of hydrogen bonds between taxanes and tubulin isotypes during the 100 ns simulation processes. For PTX, the most H-bonds are formed between PTX and βIIa-tubulin throughout the process, indicating that PTX has the stronger binding ability to βIIa-tubulin than to the other two β-tubulin isotypes. For DTX, the least H-bonds are formed between DTX and βIII-tubulin, indicating weaker binding to βIII-tubulin than to βI-tubulin and βIIa-tubulin. However, for CTX, the H-bond numbers between CTX and three β-tubulin isotypes are not so different. In other words, DTX and PTX prefer to bond βIIa-tubulin rather than bond βIII-tubulin, while CTX has less selectivity to bond to the three β-tubulin isotypes.
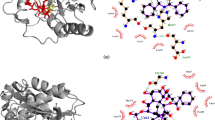
Figure 7 shows the nine equilibrium structures with the key residues after 100 ns of simulation. Figure 7a–c shows the important amino acid residues on βI-tubulin binding with PTX, DTX and CTX. The C13 side chain of DTX and CTX goes deeper into the cavity than PTX of βI-tubulin. This pattern may be related to the big size of the two hydrophobic benzene ring structures on the C13 side chain of PTX. And the main amino acids on βI-tubulin are His227, Glu276, Gln280, and Arg359. Figure 7d–f shows the binding of three taxanes with βIIa-tubulin. We observed that the orientations of three taxanes are similar, and the important binding amino acids on βIIa-tubulin are His227, Thr274, and Arg276. Figure 7g–i shows the results of three taxanes with βIII-tubulin. For different taxanes with βIII-tubulin, the orientations of them are different, and the important amino acids on βIII-tubulin are also different. Unlike the interaction with βI-tubulin and βIIa-tubulin, the C13 side chain of PTX with βIII-tubulin was partially expelled from the cavity, and the overall structure of DTX was expelled. This implies that the interactions of PTX and DTX with βIII-tubulin are weak. At the residue level, the interactions of three taxanes with βIIa-tubulin are similar, but with βIII-tubulin are quite different.

The view of the active pockets formed by the key residues (using red sticks) in surface of β-tubulin isotypes binding to taxanes (using rainbow sticks&spheres) after 100 ns of simulation. The important hydrogen bonds are represented by blue dotted lines
Based on the MD results, we carried out the MM-PBSA method (with ε = 2.0) to study the interactions of the three taxanes on the different β-tubulins (Table 6), while the decomposition of energy terms are listed in Table S5 (Supplementary Material). We also have tried different dielectric constants (ε = 4.0, 20.0), and the results are shown in Table S6 (Supplementary Material). Nevertheless, the conclusion for the relative energies is unchanged. According to the literatures [45, 46], the human body mainly consists of the βI-tubulin and βII-tubulin isotypes in early treatment, and DTX has been used in the clinic as the first-line drugs. As shown in Table 6 and Table S5 (Supplementary Material), the strong binding affinities are predicted between DTX and βI-tubulin (− 29.5 kcal/mol) or between DTX and βIIa-tubulin (− 29.0 kcal/mol), but weaker interaction between DTX and βIII-tubulin (− 17.8 kcal/mol). This may be related to the different effects of DTX for different β-tubulins. PTX has a similar situation as DTX. As for CTX, the binding affinity with βIII-tubulin (− 23.0 kcal/mol) is close to those with βI-tubulin (− 24.0 kcal/mol) and with βIIa-tubulin (− 25.9 kcal/mol). Interestingly, our theoretical modeling is consistent with the clinical studies. As it was reported, the expression of βIII-tubulin in the human body would increase after the treatment with PTX and DTX, and the resistance would occur to PTX and DTX on the later stage [32, 41]. In this case, CTX is used as the second-line drug in the clinic to prolong life, when the patient has resistance to DTX [49].
Conclusions
In this study, the interactions of three taxanes (PTX, DTX, CTX) have been investigated with different human αβ-tubulin isotypes. Initially, molecular conformations for the drugs were optimized by the DFT methods, and we obtained the low-lying conformations of CTX and their properties, such as their IR spectra and the excited states. Using homology modeling method, we successfully built the 3D models of human αβ-tubulin isotypes with high identity (> 78%) between the template and target proteins. Thus, these models are reliable for the docking and MD simulations. Next, the binding sites of αβ-tubulin dimer for CTX were found on β-tubulin by molecular docking, and these sites are similar to those for PTX and DTX. The βI-tubulin and βIIa-tubulin isotypes have substantial binding affinities docking with the taxanes.
Finally, we carried out molecular dynamics simulations for βI-tubulin, βIIa-tubulin, and βIII-tubulin with the three taxanes. Although the active pockets of different tubulin isotypes with three taxanes are comparable, the key amino acid residues and drug orientations are different. The three taxanes have quite different intermolecular actions on βIII-tubulin. The binding affinities of DTX with βI-tubulin and βIIa-tubulin are quite strong, while that on βIII-tubulin is relatively weak. On the contrary, the binding affinity of CTX on βIII-tubulin is higher than PTX and DTX. Our study supports the use of CTX as the second-line drug for the patients who are resistant to the action of DTX in the clinic. Since our computational outcomes are consistent with the clinical studies of cabazitaxel, these results should be helpful for future experimental studies of taxenes.
References
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J. Clin. 68(1):7–30. https://doi.org/10.3322/caac.21442
Yan X, Han R, Zhou J, Yu H, Yang J, Wu M (2016) Incidence, mortality and survival of female breast cancer during 2003–2011 in Jiangsu province, China. Chin J Cancer Res 28(3):321–329. https://doi.org/10.21147/j.issn.1000-9604.2016.03.06
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J (2016) Cancer statistics in China, 2015. CA Cancer J. Clin. 66(2):115–132. https://doi.org/10.3322/caac.21338
Zeng H, Chen W, Zheng R, Zhang S, Ji JS, Zou X, Xia C, Sun K, Yang Z, Li H, Wang N, Han R, Liu S, Li H, Mu H, He Y, Xu Y, Fu Z, Zhou Y, Jiang J, Yang Y, Chen J, Wei K, Fan D, Wang J, Fu F, Zhao D, Song G, Chen J, Jiang C, Zhou X, Gu X, Jin F, Li Q, Li Y, Wu T, Yan C, Dong J, Hua Z, Baade P, Bray F, Jemal A, Yu XQ, He J (2018) Changing cancer survival in China during 2003–15: a pooled analysis of 17 population-based cancer registries. Lancet Glob. Health 6(5):e555–e567. https://doi.org/10.1016/S2214-109X(18)30127-X
Chen JG, Chen HZ, Zhu J, Yang YL, Zhang YH, Huang PX, Chen YS, Zhu CY, Yang LP, Shen K, Qiang FL, Wang GR (2018) Cancer survival in patients from a hospital-based cancer registry, China. J. Cancer 9(5):851–860. https://doi.org/10.7150/jca.23039
Janke C, Bulinski JC (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12(12):773–786. https://doi.org/10.1038/nrm3227
Conde C, Caceres A (2009) Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 10(5):319–332. https://doi.org/10.1038/nrn2631
Perez EA (2009) Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 8(8):2086–2095. https://doi.org/10.1158/1535-7163.MCT-09-0366
Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo T (2000) A common pharmacophore for epothilone and taxanes: molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. 97(6):2904–2909. https://doi.org/10.1073/pnas.040546297
Kruczynski A, Barret JM, Etievant C, Colpaert F, Fahy J, Hill BT (1998) Antimitotic and tubulin-interacting properties of vinflunine, a novel fluorinated vinca alkaloid. Biochem. Pharmacol. 55(5):635–648. https://doi.org/10.1016/s0006-2952(97)00505-4
Lu Y, Chen J, Xiao M, Li W, Miller DD (2012) An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 29(11):2943–2971. https://doi.org/10.1007/s11095-012-0828-z
Jordan MA, Wilson L (2004) Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4(4):253–265. https://doi.org/10.1038/nr1317
Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M (2004) Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428(6979):198–202. https://doi.org/10.1038/nature02393
Mukhtar E, Adhami VM, Mukhtar H (2014) Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 13(2):275–284. https://doi.org/10.1158/1535-7163.mct-13-0791
Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO (2013) Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 339(6119):587–590. https://doi.org/10.1126/science.1230582
Field JJ, Diaz JF, Miller JH (2013) The binding sites of microtubule-stabilizing agents. Chem. Biol. 20(3):301–315. https://doi.org/10.1016/j.chembiol.2013.01.014
Blagosklonny MV, Fojo T (1999) Molecular effects of paclitaxel: myths and reality (a critical review). Int J Cancer 83(2):151–156. https://doi.org/10.1002/(sici)1097-0215(19991008)83:23.0.co;2-5
Lowe J, Li H, Downing KH, Nogales E (2001) Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 313(5):1045–1057. https://doi.org/10.1006/jmbi.2001.5077
Mitra A, Sept D (2008) Taxol allosterically alters the dynamics of the tubulin dimer and increases the flexibility of microtubules. Biophys. J. 95(7):3252–3258. https://doi.org/10.1529/biophysj.108.133884
Natarajan K, Senapati S (2012) Understanding the basis of drug resistance of the mutants of αβ-tubulin dimer via molecular dynamics simulations. PLoS One 7(8):e42351. https://doi.org/10.1371/journal.pone.0042351
Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E (2014) High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell 157(5):1117–1129. https://doi.org/10.1016/j.cell.2014.03.053
Kellogg EH, Hejab NMA, Howes S, Northcote P, Miller JH, Diaz JF, Downing KH, Nogales E (2017) Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-EM structures. J. Mol. Biol. 429(5):633–646. https://doi.org/10.1016/j.jmb.2017.01.001
Cao YN, Zheng LL, Wang D, Liang XX, Gao F, Zhou XL (2018) Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 143:806–828. https://doi.org/10.1016/j.ejmech.2017.11.062
Sun C, Zhu L, Zhang C, Song C, Wang C, Zhang M, Xie Y, Schaefer HF (2018) Conformers, properties, and docking mechanism of the anticancer drug docetaxel: DFT and molecular dynamics studies. J. Comput. Chem. 39(15):889–900. https://doi.org/10.1002/jcc.25165
Shah M, Dauffenbach L, Kerfoot CA, Cohen P, Culver KW, Sharma S, Wartmann M (2003) A comparative study of two novel, non-taxane microtubule stabilizing agents (XAA296A and EPO906A) utilizing the extreme drug resistance (EDR) assay in paclitaxel-sensitive or paclitaxel-resistant breast and ovarian cancers. Clin Cancer Res 9(16):6133s
Sgadari C, Toschi E, Palladino C, Barillari G, Carlei D, Cereseto A, Ciccolella C, Yarchoan R, Monini P, Sturzl M, Ensoli B (2000) Mechanism of paclitaxel activity in Kaposi’s sarcoma. J. Immunol. 165(1):509–517. https://doi.org/10.4049/jimmunol.165.1.509
Seoud MAF, Shamseddine A, Khalil AM, Charafeddine M, Geara FB (2006) Randomized trial comparing concurrent radiotherapy with cisplatin compared with paclitaxel in cervical cancer. Obstet Gynecol 107(4):43S. https://doi.org/10.1097/00006250-200604001-00101
Von Hoff DD, Goldstein D, Renschler MF (2014) Albumin-bound paclitaxel plus gemcitabine in pancreatic cancer REPLY. N. Engl. J. Med. 370(5):479–480
McGrogan BT, Gilmartin B, Carney DN, McCann A (2008) Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta 1785(2):96–132. https://doi.org/10.1016/j.bbcan.2007.10.004
Joensuu H, Bono P, Kataja V, Alanko T, Kokko R, Asola R, Utriainen T, Turpeenniemi-Hujanen T, Jyrkkio S, Moykkynen K, Helle L, Ingalsuo S, Pajunen M, Huusko M, Salminen T, Auvinen P, Leinonen H, Leinonen M, Isola J, Kellokumpu-Lehtinen PL (2009) Fluorouracil, epirubicin, and cyclophosphamide with either docetaxel or vinorelbine, with or without trastuzumab, as adjuvant treatments of breast cancer: final results of the FinHer trial. J. Clin. Oncol. 27(34):5685–5692. https://doi.org/10.1200/JCO.2008.21.4577
Gernone A, Pagliarulo A, Calderoni G, Pagliarulo V (2011) Elderly patients with metastatic castrate-resistant prostate cancer (mCRPC): safety and efficacy of docetaxel retreatment. J Clin Oncol 29(7):161. https://doi.org/10.1200/jco.2011.29.7_suppl.161
Urano N, Fujiwara Y, Doki Y, Kim SJ, Miyoshi Y, Noguchi S, Miyata H, Takiguchi S, Yasuda T, Yano M, Monden M (2006) Clinical significance of class III β-tubulin expression and its predictive value for resistance to docetaxel-based chemotherapy in gastric cancer. Int. J. Oncol. 28(2):375–381
Galsky MD, Dritselis A, Kirkpatrick P, Oh WK (2010) Cabazitaxel. Nat. Rev. Drug Discov. 9(9):677–678. https://doi.org/10.1038/nrd3254
Duran GE, Wang YC, Francisco EB, Rose JC, Martinez FJ, Coller J, Brassard D, Vrignaud P, Sikic BI (2015) Mechanisms of resistance to cabazitaxel. Mol. Cancer Ther. 14(1):193–201. https://doi.org/10.1158/1535-7163.MCT-14-0155
Paller CJ, Antonarakis ES (2011) Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther 5:117–124. https://doi.org/10.2147/DDDT.S13029
Pean E, Demolis P, Moreau A, Hemmings RJ, O'Connor D, Brown D, Shepard T, Abadie E, Pignatti F (2012) The European Medicines Agency Review of Cabazitaxel (Jevtana®) for the treatment of hormone-refractory metastatic prostate cancer: summary of the Scientific Assessment of the Committee for Medicinal Products for Human Use. Oncologist 17(4):543–549. https://doi.org/10.1634/theoncologist.2011-0364
Luduena RF (1998) Multiple forms of tubulin: different gene products and covalent modifications. Int. Rev. Cytol. 178:207–275
Santoshi S, Naik PK (2014) Molecular insight of isotypes specific β-tubulin interaction of tubulin heterodimer with noscapinoids. J. Comput. Aided Mol. Des. 28(7):751–763. https://doi.org/10.1007/s10822-014-9756-9
Derry WB, Wilson L, Khan IA, Luduena RF, Jordan MA (1997) Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified β-tubulin isotypes. Biochemistry 36(12):3554–3562. https://doi.org/10.1021/bi962724m
Yang CH, Yap EH, Xiao H, Fiser A, Horwitz SB (2016) 2-(m-Azidobenzoyl)taxol binds differentially to distinct β-tubulin isotypes. Proc. Natl. Acad. Sci. 113(40):11294–11299. https://doi.org/10.1073/pnas.1613286113
Hari M, Yang H, Zeng C, Canizales M, Cabral F (2003) Expression of class IIIβ-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil. Cytoskeleton 56(1):45–56. https://doi.org/10.1002/cm.10132
Seve P, Isaac S, Tredan O, Souquet PJ, Pacheco Y, Perol M, Lafanechere L, Penet A, Peiller EL, Dumontet C (2005) Expression of class III β-tubulin is predictive of patient outcome in patients with non-small cell lung cancer receiving vinorelbine-based chemotherapy. Clin. Cancer Res. 11(15):5481–5486. https://doi.org/10.1158/1078-0432.CCR-05-0285
Ferrandina G, Martinelli E, Zannoni GF, Distefano M, Paglia A, Ferlini C, Scambia G (2007) Expression of class III β-tubulin in cervical cancer patients administered preoperative radiochemotherapy: correlation with response to treatment and clinical outcome. Gynecol. Oncol. 104(2):326–330. https://doi.org/10.1016/j.ygyno.2006.08.046
Sève P, Dumontet C (2008) Is class III β-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol 9(2):168–175. https://doi.org/10.1016/s1470-2045(08)70029-9
Katsetos CD, Herman MM, Mork SJ (2003) Class III β-tubulin in human development and cancer. Cell Motil. Cytoskeleton 55(2):77–96. https://doi.org/10.1002/cm.10116
Ferrandina G, Zannoni GF, Martinelli E, Paglia A, Gallotta V, Mozzetti S, Scambia G, Ferlini C (2006) Class III β-tubulin overexpression is a marker of poor clinical outcome in advanced ovarian cancer patients. Clin. Cancer Res. 12(9):2774–2779. https://doi.org/10.1158/1078-0432.ccr-05-2715
Magnani M, Ortuso F, Soro S, Alcaro S, Tramontano A, Botta M (2006) The βI/βIII-tubulin isoforms and their complexes with antimitotic agents. FEBS J. 273(14):3301–3310. https://doi.org/10.1111/j.1742-4658.2006.05340.x
Akasaka K, Maesawa C, Shibazaki M, Maeda F, Takahashi K, Akasaka T, Masuda T (2009) Loss of class III β-tubulin induced by histone deacetylation is associated with chemosensitivity to paclitaxel in malignant melanoma cells. J. Investig. Dermatol. 129(6):1516–1526. https://doi.org/10.1038/jid.2008.406
Smiyun G, Azarenko O, Miller H, Rifkind A, LaPointe NE, Wilson L, Jordan MA (2017) βIII-tubulin enhances efficacy of cabazitaxel as compared with docetaxel. Cancer Chemother. Pharmacol. 80(1):151–164. https://doi.org/10.1007/s00280-017-3345-2
Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV (2003) Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr. Cancer Drug Targets 3(1):1–19. https://doi.org/10.2174/1568009033333754
Ferlini C, Raspaglio G, Cicchillitti L, Mozzetti S, Prislei S, Bartollino S, Scambia G (2007) Looking at drug resistance mechanisms for microtubule interacting drugs: does TUBB3 work? Curr. Cancer Drug Targets 7(8):704–712. https://doi.org/10.2174/156800907783220453
Sissung TM, Baum CE, Deeken J, Price DK, Aragon-Ching J, Steinberg SM, Dahut W, Sparreboom A, Figg WD (2008) ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin. Cancer Res. 14(14):4543–4549. https://doi.org/10.1158/1078-0432.ccr-07-4230
Kavallaris M (2010) Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 10(3):194–204. https://doi.org/10.1038/nrc2803
Kulkarni SA, Feng SS (2011) Effects of surface modification on delivery efficiency of biodegradable nanoparticles across the blood-brain barrier. Nanomedicine 6(2):377–394. https://doi.org/10.2217/nnm.10.131
Culine S (2018) Refining the use of cabazitaxel in metastatic castrate-resistant prostate cancer. Eur. J. Cancer 97:30–32. https://doi.org/10.1016/j.ejca.2018.04.002
Zhang ML, Song C, Yao Z, Ji Q (2012) Theoretical studies of the structure and properties of anticancer drug taxol. Curr Org Chem 16(19):2321–2331. https://doi.org/10.2174/138527212803520281
Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. BMC Biol. 9:71. https://doi.org/10.1186/1741-7007-9-71
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr. JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16 Rev. B.01. Wallingford, CT
Cavasotto CN, Phatak SS (2009) Homology modeling in drug discovery: current trends and applications. Drug Discov. Today 14(13–14):676–683. https://doi.org/10.1016/j.drudis.2009.04.006
The UniProt C (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45(D1):D158–D169. https://doi.org/10.1093/nar/gkw1099
McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R (2013) Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 41:W597–W600. https://doi.org/10.1093/nar/gkt376
Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30:S162–S173. https://doi.org/10.1002/elps.200900140
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics (Oxford, England) 27(3):343–350. https://doi.org/10.1093/bioinformatics/btq662
Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, Schwede T (2017) The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. 45(D1):D313–D319. https://doi.org/10.1093/nar/gkw1132
Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 7(1):10480. https://doi.org/10.1038/s41598-017-09654-8
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1):W296–W303. https://doi.org/10.1093/nar/gky427
Gilson MK, Zhou HX (2007) Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. 36:21–42. https://doi.org/10.1146/annurev.biophys.36.040306.132550
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30(16):2785–2791. https://doi.org/10.1002/jcc.21256
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26(16):1701–1718. https://doi.org/10.1002/jcc.20291
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J. Comput. Chem. 25(9):1157–1174. https://doi.org/10.1002/jcc.20035
Leontyev IV, Stuchebrukhov AA (2010) Electronic polarizability and the effective pair potentials of water. J. Chem. Theory Comput. 6(10):3153–3161. https://doi.org/10.1021/ct1002048
Hess B (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4(1):116–122. https://doi.org/10.1021/ct700200b
Mozzetti S, Ferlini C, Concolino P, Filippetti F, Raspaglio G, Prislei S, Gallo D, Martinelli E, Ranelletti FO, Ferrandina G, Scambia G (2005) Class III β-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 11(1):298–305
Seve P, Mackey J, Isaac S, Tredan O, Souquet PJ, Perol M, Lai R, Voloch A, Dumontet C (2005) Class III β-tubulin expression in tumor cells predicts response and outcome in patients with non-small cell lung cancer receiving paclitaxel. Mol. Cancer Ther. 4(12):2001–2007. https://doi.org/10.1158/1535-7163.MCT-05-0244
Gan PP, Pasquier E, Kavallaris M (2007) Class III β-tubulin mediates sensitivity to chemotherapeutic drugs in non-small cell lung cancer. Cancer Res. 67(19):9356–9363. https://doi.org/10.1158/0008-5472.CAN-07-0509
Guo J, Wang X, Sun H, Liu H, Yao X (2012) The molecular basis of IGF-II/IGF2R recognition: a combined molecular dynamics simulation, free-energy calculation and computational alanine scanning study. J. Mol. Model. 18(4):1421–1430. https://doi.org/10.1007/s00894-011-1159-4
Jing X, Xiaoqiang H, Yushan Z (2019) Using molecular dynamics simulations to evaluate active designs of cephradine hydrolase by molecular mechanics/Poisson – Boltzmann surface area and molecular mechanics/generalized born surface area methods. RSC Adv. 9:13868–13877. https://doi.org/10.1039/c9ra02406a
Kumari R, Kumar R, Open Source Drug Discovery C, Lynn A (2014) g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54(7):1951–1962. https://doi.org/10.1021/ci500020m
Stepanian SG, Reva ID, Radchenko ED, Adamowicz L (1998) Conformational behavior of α-alanine. Matrix-isolation infrared and theoretical DFT and ab initio study. J Phys Chem A 102(24):4623–4629. https://doi.org/10.1021/jp973479z
Yu D, Kang W, Hao F, Li Z (2017) Spectroscopic studies and structural elucidation of cabazitaxel. Chinese J Magn Reson 34(2):191–199
Baker D, Sali A (2001) Protein structure prediction and structural genomics. Science 294(5540):93–96. https://doi.org/10.1126/science.1065659
Bowie JU, Luthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253(5016):164–170
Luthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356(6364):83–85. https://doi.org/10.1038/356083a0
Kumbhar BV, Borogaon A, Panda D, Kunwar A (2016) Exploring the origin of differential binding affinities of human tubulin isotypes αβII, αβIII and αβIV for DAMA-colchicine using homology modelling, molecular docking and molecular dynamics simulations. PLoS One 11(5):e0156048. https://doi.org/10.1371/journal.pone.0156048
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants No. 10904111, 11604238, and 21772146), the Tianjin Natural Science Foundation (11JCYBJC14500), the Science & Technology Development Fund of Tianjin Education Commission for Higher Education (2019KJ175), and the China Postdoctoral Science Foundation (20100470792). We appreciate the supply of the computing resources by USTC. The research at the University of Georgia was supported by the National Science Foundation (Grant CHE-1661604). The visit of MZ to the Center for Computational Quantum Chemistry, the University of Georgia was very helpful.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 11727 kb)
Rights and permissions
About this article

Cite this article
Zhu, L., Zhang, C., Lü, X. et al. Binding modes of cabazitaxel with the different human β-tubulin isotypes: DFT and MD studies. J Mol Model 26, 162 (2020). https://doi.org/10.1007/s00894-020-04400-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04400-w