
Abstract
Cabazitaxel is a novel taxane approved for treatment of metastatic hormone-refractory prostate cancer in patients pretreated with docetaxel. Cabazitaxel, docetaxel, and paclitaxel bind specifically to tubulin in microtubules, disrupting functions essential to tumor growth. High levels of βIII-tubulin isotype expression are associated with tumor aggressivity and drug resistance. To understand cabazitaxel’s increased efficacy, we examined binding of radio-labeled cabazitaxel and docetaxel to microtubules and the drugs’ suppression of microtubule dynamic instability in vitro in microtubules assembled from purified bovine brain tubulin containing or devoid of βIII-tubulin. We found that cabazitaxel suppresses microtubule dynamic instability significantly more potently in the presence of βIII-tubulin than in its absence. In contrast, docetaxel showed no βIII-tubulin-enhanced microtubule stabilization. We also asked if the selective potency of cabazitaxel on βIII-tubulin-containing purified microtubules in vitro extends to cabazitaxel’s effects in human tumor cells. Using MCF7 human breast adenocarcinoma cells, we found that cabazitaxel also suppressed microtubule shortening rates, shortening lengths, and dynamicity significantly more strongly in cells with normal levels of βIII-tubulin than after 50% reduction of βIII-tubulin expression by siRNA knockdown. Cabazitaxel also more strongly induced mitotic arrest in MCF7 cells with normal βIII-tubulin levels than after βIII-tubulin reduction. In contrast, docetaxel had little or no βIII-tubulin-dependent selective effect on microtubule dynamics or mitotic arrest. The selective potency of cabazitaxel on purified βIII-tubulin-containing microtubules and in cells expressing βIII-tubulin suggests that cabazitaxel may be unusual among microtubule-targeted drugs in its superior anti-tumor efficacy in tumors overexpressing βIII-tubulin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cabazitaxel (Jevtana®) and docetaxel (Taxotere®) (Fig. 1a) are semisynthetic analogs of paclitaxel that are effective anticancer agents in the clinic. Cabazitaxel is a microtubule-targeted cancer chemotherapeutic agent that has been approved clinically for treatment of metastatic castration-resistant prostate cancer after failure of docetaxel treatment [1, 2].

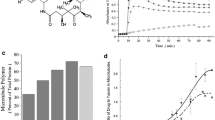
a Structures of cabazitaxel and docetaxel. b Binding of cabazitaxel to unfractionated or βIII-tubulin-depleted microtubules. Tubulin was incubated in the presence of [14C]-cabazitaxel to form microtubules, and binding affinity and stoichiometry were determined. Cabazitaxel bound to unfractionated (closed symbols, four independent experiments) and βIII-tubulin-depleted microtubules (open symbols, two independent experiments) with a K D of 7.4 ± 0.9 (SD) μmol/L and 8.3 ± 1.2 (SD) μmol/L, respectively. c Binding of docetaxel to unfractionated or βIII-tubulin-depleted microtubules. Tubulin was incubated in the presence of [14C]-docetaxel to form microtubules, and binding affinity and stoichiometry were determined. Docetaxel bound to unfractionated (closed symbols, four independent experiments) and βIII-tubulin-depleted microtubules (open symbols, two independent experiments) with a K D of 6.8 ± 0.2 (SD) μmol/L and 7.9 ± 0.3(SD) μmol/L, respectively
Microtubules are dynamic tubular cytoskeletal filaments composed of tubulin, which is a heterodimer of α and β tubulin monomers. Like other taxanes (paclitaxel and docetaxel), cabazitaxel enhances microtubule assembly, arrests cells in mitosis, and suppresses microtubule dynamic instability, a behavior that involves frequent transitions between slow growth and rapid shortening of individual microtubules [3,4,5]. Suppression of microtubule dynamic instability can prevent cells from completing mitosis, resulting in inhibition of cell proliferation and ultimately in cell death. Suppression of dynamic instability also impairs essential non-mitotic functions of cancer cells including migration, signaling, intracellular trafficking and secretion [6, 7].
The seven identified isotypes of β-tubulin are expressed differently in different cell types [8, 9]. The βIII-tubulin isotype is predominantly expressed in neurons and Sertoli cells of the testis. Its expression is often associated with drug-resistant and aggressive cancers [10,11,12,13,14,15]. The detailed mechanistic effects of βIII-tubulin levels on cabazitaxel’s and docetaxel’s actions in vitro and in cells have not been examined.
We compared the binding of cabazitaxel and docetaxel to microtubules and their interactions in vitro with purified microtubules either containing or depleted of βIII-tubulin. We found that in vitro both drugs bind to microtubules with similar K D’s whether the microtubules contain βIII-tubulin or are depleted of it. However, cabazitaxel’s suppressive effects on microtubule dynamic instability were greater in the presence of βIII-tubulin than in its absence whereas the suppressive effects of docetaxel were independent of the level of βIII-tubulin isotype. We examined the drugs’ effects in MCF7 cells before and after βIII-tubulin siRNA knockdown and found that reduction of βIII-tubulin levels significantly reduced mitotic arrest and reduced suppression of dynamic instability by cabazitaxel. In contrast, docetaxel’s effects in cells were minimally altered by reduction of βIII-tubulin. The results suggest that cabazitaxel’s superior efficacy in tumors resistant to other taxanes results, at least in part, from its enhanced inhibition of the dynamic instability of microtubules that contain high levels of βIII-tubulin.
Materials and methods
Chemicals and other materials were purchased from Sigma-Aldrich, LLC (St. Louis, MO, USA) unless otherwise noted. Cabazitaxel and docetaxel were provided by Sanofi-Oncology (Cambridge, MA, USA), dissolved in vehicle dimethylsulfoxide (DMSO) (ATCC, Manassas, VA, USA) and stored as 10 mmol/L aliquots at −20 °C.
Tubulin purification, βIII-tubulin depletion, microtubule assembly and length determinations
Bovine brain tubulin was purified by cycles of temperature- and GTP-dependent polymerization and depolymerization followed by phosphocellulose chromatography and depletion of βIII-tubulin in vitro [16]. Anti-βIII-tubulin monoclonal antibodies were generated by growing TUJ1-expressing human-mouse hybridoma cells (gift of Anthony Frankfurter and Anthony J. Spano, University of Virginia). Western blots using the TUJ1 antibody and the tubulin used in these experiments are shown [17] and indicate complete absence of βIII-tubulin. Microtubules were assembled, and their lengths and concentrations in suspension were calculated from length distributions obtained by electron microscopy and polymer mass determinations [16].
Drug binding to microtubules composed of unfractionated or βIII-depleted tubulin
Unfractionated or βIII-depleted tubulin (20 µmol/L) was polymerized (60 min) in the presence of [14C]-cabazitaxel or [14C]-docetaxel (5–40 µmol/L; specific activity 85 mCi/mmol). Microtubules were collected by centrifugation through glycerol/DMSO cushions and bound [14C]-drug and binding stoichiometries determined [17]. Four and two independent experiments were performed with unfractionated and βIII-isotype-depleted tubulin, respectively.
Microtubule dynamic instability in vitro
Drug effects (100 nmol/L) on dynamic instability parameters at plus ends of steady-state microtubules assembled with unfractionated or βIII-depleted tubulin were determined by differential interference contrast video microscopy [18]. Parameters included growth rates and lengths, shortening rates and lengths, transition frequencies from the growth or attenuated (paused) state to shortening (termed catastrophe), transition frequencies from shortening to the growth or paused state (termed rescue), and the overall dynamicity of the microtubules (the total visually measurable growth and shortening of the microtubules per unit time). Between 27 and 42 microtubules were analyzed per condition in at least three independent experiments, and data pooled for statistical analysis.
Statistical analysis was carried out in R [19,20,21] by two-way ANOVA with or without transformation (Supplementary Methods). Graphs were made in R using ggplot2 [22] and detailed in Adobe Illustrator. Significance notations indicate comparison to the appropriate vehicle control unless otherwise noted. Data that were transformed prior to analysis are presented in the original scale as the back-transformed mean and 95% confidence levels. Raw data are presented as mean and SEM in Table 1.
Cell culture
MCF7 human breast adenocarcinoma cells (ATCC, Manassas, VA, USA) expressing green fluorescence EGFP-α-tubulin were used due to their flatness which enables determination of dynamic instability in living cells [5]. The EGFP-α-tubulin-expressing cells were indistinguishable from unmodified cells except for their fluorescent microtubules and their doubling time of 35 h, which was 20% slower than unmodified MCF7 cells.
βIII-tubulin knockdown in MCF7 cells
Cells were transfected with a βIII-tubulin siRNA pool (50 µmol/L) prepared from three individual siRNAs at a ratio of 1:1:1 according to the manufacturer’s guidelines (Thermo Fisher Scientific, Waltham, MA, USA) or control siRNA at a final concentration of 10 nmol/L [17]. Mock transfection was performed with OPTI-MEM containing no siRNA. Cells (100,000 cells/mL) were incubated 24 h with transfection mixture (10 nmol/L control siRNA or βIII-tubulin siRNA or mock transfection reagent). Media was then replaced with fresh media containing 10% FBS. For live cell imaging, control and βIII-tubulin siRNA-transfection mixtures included 10 nmol/L BLOCK-iT Alexa Fluor Red Fluorescent Oligo (Invitrogen, Grand Island, NY, USA) to distinguish siRNA-transfected cells.
Cell proliferation
Transfected cells were seeded (100,000 cells/mL) in 96-well plates in triplicate (100 µL/well). At 48-, 96- or 144 h-post knockdown, cells were fixed with 10% (w/v) trichloroacetic acid (TCA) (Thermo Fisher Scientific, Waltham, MA, USA), stained with 0.4% (w/v) sulforhodamine B (SRB) and absorbance read at 490 nm. Experiments were repeated four times.
Effects of cabazitaxel or docetaxel on cell proliferation were also measured by counting cells by hemocytometer. Cells were transfected and seeded (100,000 cells/mL) in 12-well plates (500 µL/well) for 24 h to allow cell attachment and recovery. Medium with drug (0.03–3 nmol/L) was added for an additional 72 h, and cells were harvested and counted. Results are means of 4–5 experiments.
Cell cycle determination by flow cytometry
Cell cycle analysis was determined 48 and 96 h after transfection without the drugs. Cells were harvested (adherent and suspended), fixed in 70% ethanol for 24 h at 4 °C, stained with propidium iodide (EMD Millipore, Billerica, MA, USA), analyzed using Guava EasyCyte flow cytometer (Guava Technologies, Inc.) (2000 events per condition) and ModFitLT software (Verity Software House Inc., Topsham, ME, USA).
Mitotic arrest
Following the 48-h βIII-tubulin knockdown, cells were seeded at 100,000 cells/mL in 6-well plates for 24 h. Cabazitaxel or docetaxel (3–100 nmol/L) was added for an additional 20 h. Adherent and detached cells were harvested, fixed (3.7% formaldehyde), stained with Alexa Fluor-594-conjugated anti-phosphohistone H3 antibody (Cell Signaling Technology, Danvers, MA, USA), and mounted onto glass with ProLong Gold DAPI (4′,6-diamidino-2-phenylindole) antifade reagent (Invitrogen, Grand Island, NY, USA). Percentages of mitotic cells were determined by counting cells containing condensed DNA that were positive for phosphohistone H3 (a mitotic marker) and dividing by total cell number. Five hundred cells were counted per condition in each of 4 independent experiments. IC50’s for mitotic arrest were calculated and significance determined using Student’s t test.
Immunofluorescence microscopy
Cells were grown on poly-l-lysine-coated coverslips (48 h), incubated with 10 nmol/L cabazitaxel or docetaxel (20 h), and fixed in 3.7% formaldehyde (20 min, 37 °C) followed by methanol (10 min, −20 °C) and stained [5] and imaged using the same exposure for each sample with an Olympus FLV1000S Spectral laser scanning confocal microscope (Olympus America Inc., Central Valley, PA, USA).
Microtubule dynamic instability in cells
Control or βIII-knockdown MCF7 cells were seeded onto glass coverslips pre-coated with 50 μg/mL poly-l-lysine, 10 μg/mL laminin, and 20 μg/mL fibronectin (2 h, 37 °C). To enhance flattening, cells were incubated for 24 h with 1% FBS in DMEM. Forty-eight hours after transfection cells were incubated with drug (10 nmol/L) for 20 h (the time required for equivalent uptake of cabazitaxel and docetaxel into cells [5]). Time-lapse images of microtubules were acquired with a 100× objective on a Nikon Eclipse E800 microscope with a Hamamatsu Orca II digital camera (Hamamatsu, Japan), driven by Metamorph software (Molecular Devices, Sunnyvale, CA, USA) [5, 17]. The plus ends of microtubules were tracked, graphed as a function of time and analyzed in MT-LHAP v.3, r.1 (developed by Emin Oroudjev, http://www.igorexchange.com/node/1767). Increase or decrease in length of ≥0.3 μm between two points were considered growth or shortening events. Changes in length of <0.3 μm were called periods of “attenuated dynamics” or “pause”. Dynamicity is the sum of total lengths grown and shortened divided by total duration of imaging a microtubule. Results are from >50 individual microtubules from ≥10 or more cells for each condition, and from at least three experiments for each condition. Data were pooled prior to statistical analysis, as for in vitro data (see above and Supplementary Methods). Raw mean and SEM are shown in Table 2.
Results
In vitro
Binding of cabazitaxel to microtubules assembled from unfractionated tubulin or from βIII-depleted tubulin
Cabazitaxel and docetaxel (Fig. 1a) are microtubule stabilizing drugs that at relatively high concentrations induce microtubule polymerization [5]. To test drug binding to microtubules assembled from unfractionated and βIII-depleted tubulin, we purified tubulin from bovine brain (which contains 25% βIII-tubulin) and depleted the βIII-tubulin to undetectable levels by immunoaffinity chromatography; the absence of βIII-tubulin is shown in Lopus et al. [17], which shows western blots of the isotype content in the same tubulin preparation. We then determined the binding affinity and stoichiometry of [14C]-cabazitaxel to microtubules assembled in vitro from tubulin in the presence or absence of the βIII-tubulin isotype. [14C]-cabazitaxel bound to the microtubules in a concentration-dependent manner (Fig. 1b). At low cabazitaxel concentrations (≤10 μmol/L) binding to microtubules assembled from unfractionated or βIII-depleted tubulin was similar. At 20 μmol/L cabazitaxel, 1 molecule of cabazitaxel bound per molecule of tubulin in microtubules composed of unfractionated tubulin, and 1.25 molecules of cabazitaxel bound per tubulin molecule in microtubules composed of βIII-tubulin-depleted tubulin (not a statistically significant difference). K Ds were calculated as the IC50 for binding at the lowest and highest cabazitaxel concentrations tested with unfractionated or βIII-depleted microtubules. Cabazitaxel is bound to unfractionated microtubules with K D 7.4 ± 0.9 (SD) µmol/L and to βIII-depleted microtubules with K D 8.3 ± 1.2 (SD) µmol/L, presumably at the taxane binding site. The 1.1-fold difference in K Ds was not statistically significant. Polymer mass levels were approximately the same with either unfractionated or βIII-depleted tubulin at all drug concentrations (data not shown). In conclusion, the binding of cabazitaxel to tubulin was independent of βIII-tubulin content (Fig. 1b).
Binding of docetaxel to microtubules assembled from unfractionated or βIII-depleted tubulin
Experiments to determine [14C]-docetaxel binding were performed concurrently with those for [14C]-cabazitaxel binding. [14C]-docetaxel bound to microtubules in a concentration-dependent manner similar to [14C]-cabazitaxel (Fig. 1c). The K Ds for [14C]-docetaxel’s binding to unfractionated and βIII-depleted microtubules were 6.8 ± 0.2 (SD) µmol/L and 7.9 ± 0.3 (SD) µmol/L, respectively. The 1.2-fold difference between the K Ds was not statistically significant. At one of the highest concentrations tested (20 µmol/L), approximately one molecule of [14C]-docetaxel bound per molecule of tubulin dimer in microtubules assembled from either unfractionated or βIII-depleted tubulin. Thus, microtubule binding of docetaxel is independent of βIII-tubulin content.
Comparison of βIII-tubulin effects on dynamic instability suppression in vitro by cabazitaxel and docetaxel
Microtubules assembled from purified tubulin display periods of relatively slow growth and fast shortening, with infrequent switching between growth and shortening. Microtubules spend an appreciable amount of time neither growing nor shortening detectably (called “attenuation” or “pause”). This GTP-dependent nonequilibrium behavior is called dynamic instability [3]. We examined the in vitro effects of cabazitaxel and docetaxel on dynamic instability parameters of microtubules containing or depleted of βIII-tubulin (Table 1).
In the present study, in the absence of drug, removal of βIII-tubulin had no detectable effects on the dynamic instability parameters in vitro of the remaining mixture of isotypes. Microtubules made from unfractionated and βIII-depleted tubulin microtubules grew at indistinguishable rates (2.0 ± 0.1 and 1.9 ± 0.1 μm/min, respectively), shortened at similar rates (29.3 ± 1.4 and 28.4 ± 1.8 μm/min, respectively), and had overall dynamicities of 2.4 and 2.5 μm/min for unfractionated and βIII-depleted microtubules, respectively (Table 1). These results may seem surprising because when previously analyzed separately [23] microtubules composed solely of purified βIII-tubulin were approximately two-fold more dynamic than microtubules made from either the purified βII-isotype or the βIV-isotype; thus one might expect that depletion of βIII-tubulin should slow the dynamics of the remaining mixture. However, we found that microtubules made from a 50:50 mixture of βIII- and βII-tubulin did not exhibit dynamics in between the two isotypes, but rather, the microtubules made from 50% purified βII-tubulin and 50% purified βIII-tubulin exhibited dynamics that were similar to those of 100% βII tubulin. Moreover, a mixture of 20% βIII-tubulin and 80% βII-tubulin exhibited dynamics that were slower than microtubules made from 100% βII-tubulin. Thus, changing the ratios of the various β-tubulin isotypes results in unpredictable effects on dynamics of the resulting mixture (discussed in [23]).
Depletion of βIII-tubulin reduces the effects of cabazitaxel, but not docetaxel, on microtubule dynamics in vitro
Cabazitaxel suppressed microtubule dynamic instability more when βIII-tubulin levels were high (i.e., “unfractionated” tubulin) (Table 1). Specifically, 100 nmol/L cabazitaxel suppressed shortening rates of unfractionated microtubules and βIII-depleted microtubules, by 49 and 31%, respectively, shortening length by 50 and 23%, respectively, and overall dynamicity by 49 and 33%, respectively. Cabazitaxel increased rescue frequency by 90 and 69%, respectively. In contrast, docetaxel suppressed dynamic instability independent of the microtubule content of βIII-tubulin. Specifically, 100 nmol/L docetaxel suppressed shortening rates of unfractionated microtubules and βIII-depleted microtubules, by 48 and 65%, respectively, shortening length by 49 and 50%, respectively, and overall dynamicity by 56 and 50%, respectively.
To determine whether or not these differences observed in the presence and absence of βIII-tubulin were statistically significant, we performed a two-way ANOVA (see Methods). Data were transformed prior to analysis to meet the assumptions of the ANOVA, and are presented as the back-transformed means in the original scale with error bars showing 95% confidence levels (Fig. 2 and Supplementary Fig. 1; raw data are in Table 1). For shortening length (Fig. 2a), the ANOVA indicated a significant interaction (p < 0.01) between drug treatment (vehicle control, docetaxel, cabazitaxel) and βIII-tubulin content (unfractionated vs. βIII-depleted). Specifically, cabazitaxel suppressed shortening length significantly less potently in βIII-depleted microtubules than in unfractionated microtubules (p < 0.001). No such differences were observed for docetaxel or the vehicle controls. Furthermore, while docetaxel and cabazitaxel were similarly potent at suppressing shortening length in unfractionated microtubules, cabazitaxel was significantly less potent than docetaxel in βIII-depleted microtubules (p < 0.001).

Bar graphs showing effects of vehicle (veh, clear bars), 100 nmol/L docetaxel (doc, light gray bars) and 100 nmol/L cabazitaxel (cbz, dark gray bars) on major parameters of dynamic instability in microtubules assembled in vitro with unfractionated bovine brain tubulin (“Unfractionated”) or with bovine brain tubulin depleted of βIII-tubulin (“βIII-depleted”). a–c Show shortening length, shortening rate, and dynamicity, respectively. Data were transformed to meet the assumptions of two-way ANOVA, and all statistical analyses were carried out on the transformed scale. Graphs show means and 95% confidence levels back-transformed to the original scale. In cases where the two-way ANOVA indicated a significant interaction between drug (veh, doc, cbz) and tubulin type, subsequent pairwise comparisons were carried out using Tukey’s correction for multiple comparisons. Bars are arranged side-by-side to allow comparison of unfractionated and βIII-depleted tubulin. Asterisks indicate statistical significance of difference at *p < 0.05, **p < 0.01, ***p < 0.001 levels, respectively, relative to the appropriate vehicle control or as indicated by lines
Shortening rate (Fig. 2b) followed the same pattern, although the ANOVA did not reach the significant cutoff of p ≤ 0.05 (p = 0.054). As with shortening length, cabazitaxel was less potent in βIII-depleted microtubules than in unfractionated microtubules. Furthermore, cabazitaxel and docetaxel suppressed shortening rate with similar potency in unfractionated microtubules, whereas in βIII-depleted microtubules the effects of cabazitaxel were less potent than docetaxel. Overall dynamicity (Fig. 2c) also followed the same pattern (ANOVA p < 0.05). The difference between cabazitaxel in unfractionated vs. βIII-depleted microtubules did not reach significance. However, while the drugs were similarly potent in unfractionated microtubules, in the case of βIII-depleted microtubules, cabazitaxel was significantly less potent than docetaxel (p < 0.01). Taken together, these results support the conclusion that the presence of the βIII-tubulin isotype in microtubules enhances the suppressive effects of cabazitaxel on dynamic instability.
In cells
Comparison of cabazitaxel and docetaxel effects in cells with and without βIII-tubulin isotype knockdown
βIII-tubulin levels are important in tumor aggressivity and drug resistance [14, 15]; thus we extended our observations to the effects of altering cellular levels of βIII-tubulin. βIII-tubulin expression was reduced in MCF7 human breast cancer cells with βIII-tubulin siRNA as determined by Western blotting [17], and the drug effects were compared with effects in cells with unaltered βIII-tubulin expression transfected with either control siRNA or mock-transfected (“Materials and Methods”). MCF7 cells were among the four highest βIII-tubulin expressors in a comparison of 12 human cell lines [24]. As we reported previously [17], 48 and 96 h after knockdown, βIII-tubulin expression in MCF7 cells was reduced by 45 and 60%, respectively, from 3 to 5% to ~2% of total tubulin. When βIII-tubulin expression was reduced below 2%, the cells invariably died (data not shown).
Forty-eight and 96-h post-transfection, cells with reduced βIII-tubulin expression and no drug proliferated at the same rate as control-transfected cells. However, >96 h after transfection, proliferation of the βIII-tubulin knockdown cells markedly slowed (Fig. 3a). At 48 and 96 h control and βIII-tubulin knockdown cells had similar proportions of cells in G0/G1, S and G2/M phases (Fig. 3b). Thus, we confined our experiments to the first 96 h after βIII-tubulin knockdown.

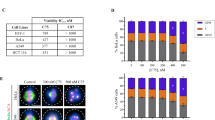
a Effects of βIII-tubulin siRNA knockdown on MCF7 cell proliferation in the absence of drug measured by sulforhodamine B assay. Cell proliferation was not significantly affected by βIII-tubulin knockdown for the first 96 h after transfection. After the 96-h proliferation was significantly reduced by βIII-tubulin knockdown (open squares), in contrast to mock transfection (open diamonds), and control transfection (filled squares), thus limiting the duration of experiments. b Cell cycle distribution after 48 h (top row of panels) and 96 h (bottom row of panels) of βIII-tubulin knockdown as determined by flow cytometry. Control siRNA-transfected cells are shown in the panels on the left and results of βIII-tubulin knockdown are shown in the panels on the right. Each figure shows the mean percentages of cells in sub-G1, G1/G0, S and G2/M phases, ± SEM of three experiments. For 96 h, βIII-tubulin knockdown had no effects on cell cycle transit, but after 96-h cell cycle transit was inhibited (data not shown); thus experiments on microtubule behavior were confined to the first 96 h after knockdown. c, d In the presence of normal levels of βIII-tubulin, mitotic arrest by cabazitaxel (c) was significantly greater in control-transfected cells (filled squares) than after βIII-tubulin knockdown (open squares). As shown in d, βIII-tubulin knockdown had no statistically significant effect on docetaxel-induced mitotic arrest. Each data point is the average of four independent assays. e Cabazitaxel inhibited cell proliferation 10 times more potently than docetaxel, but there was no effect of βIII-tubulin knockdown on inhibition of proliferation by either drug. 24 h after knockdown cells were incubated with drug for 72 h and cell number was determined by counting by hemocytometer
βIII-tubulin knockdown had no significant effect on mitotic arrest by docetaxel, but significantly reduced mitotic arrest by cabazitaxel
Forty-eight hours after βIII-tubulin knockdown (when βIII-tubulin expression was reduced by 45% [17]), control-transfected and βIII-tubulin knockdown cells were incubated with a range of drug concentrations for an additional 20 h. Cabazitaxel (Fig. 3c) and docetaxel (Fig. 3d) arrested both control and βIII-tubulin knockdown cells in mitosis. Mitotic arrest IC50 for cabazitaxel was significantly more potent in control-transfected cells (8 nmol/L ± 2.1 SEM) than in cells with reduced βIII-tubulin (18 nmol/L ± 1.7 SEM, a 2.3-fold difference, Fig. 3c, p < 0.01). In contrast, the difference was not statistically significant for docetaxel (IC50 ± SEM, 7.8 nmol/L ± 2.2 for control-transfected cells and 10 nmol/L ± 1.4, respectively, Fig. 3d).
The antiproliferative potencies of cabazitaxel and docetaxel in MCF7 cells were not detectably altered by βIII-tubulin reduction
The effects of cabazitaxel and docetaxel on proliferation of MCF7 cells were determined after a 72 h drug incubation following a 48 h βIII-tubulin knockdown (Fig. 3e). Inhibition of proliferation by cabazitaxel was similar with or without βIII-tubulin knockdown (IC50 ± SEM: 0.09 ± 0.01, and 0.18 ± 0.09 nmol/L, respectively). In agreement with the results of Azarenko et al. [5] inhibition of proliferation by docetaxel was tenfold less potent than with cabazitaxel (Fig. 3e), but it was similarly unaltered by βIII-tubulin reduction (IC50 ± SEM: 1.2 ± 0.2 and 1.1 ± 0.2 nmol/L).
Cabazitaxel and docetaxel-induced similar changes in microtubule organization regardless of βIII-tubulin content
MCF7 cells were transfected with siRNA (either control or βIII-tubulin knockdown), incubated with 10 nmol/L cabazitaxel or docetaxel for 20 h, then fixed and stained to examine microtubule organization by immunofluorescence microscopy (Fig. 4). In the absence of drug, cells with reduced βIII-tubulin levels had similar morphology to those of control-transfected cells, both in mitosis and in interphase (Fig. 4, top row). The presence of βIII-tubulin is indicated by the red stain in the left two columns. The red stain is reduced or absent after βIII-tubulin knockdown (two columns on right). In the absence of drug (top row), both with and without βIII-tubulin knockdown, mitotic cells appeared normal with condensed chromosomes in compact bipolar spindles. Mitotically arrested cells induced with either 10 nmol/L cabazitaxel or 10 nmol/L docetaxel (20 h) contained spindles typical of cells treated with microtubule-targeted drugs, both after control siRNA-transfection and after βIII-tubulin knockdown; cabazitaxel and docetaxel-induced spindles with uncongressed chromosomes and were monopolar and ball-shaped or multiastral or bipolar. Both drugs induced microtubule bundles typical of taxane treatment in interphase of both control-transfected cells and after βIII-tubulin knockdown.

Immunofluorescence microscopy of MCF7 cells in interphase and mitosis showing the effects of cabazitaxel and docetaxel on microtubule organization after control transfection (left two columns ) and after βIII-tubulin knockdown (right two columns). All interphase cell images were taken at one exposure and all mitotic cell images were taken at one exposure. There were no major differences in the morphology of control-transfected and βIII-tubulin knockdown cells in the absence of drug in mitosis or interphase. Both cabazitaxel and docetaxel disrupted mitotic spindles and induced microtubule bundling in interphase. α-Tubulin green, βIII-tubulin red, DNA blue. Scale bars are 5 μm (large) and 10 μm (small)
Cabazitaxel, but not docetaxel, inhibited microtubule dynamic instability significantly more potently in MCF7 cells when βIII-tubulin was present at normal levels than after its reduction
We examined the effects of βIII-tubulin knockdown on suppression of microtubule dynamic instability parameters by cabazitaxel and docetaxel in living MCF7 cells in interphase (Table 2). In the absence of cabazitaxel or docetaxel, reduction of βIII-tubulin had no significant effect on dynamic instability. After siRNA knockdown of βIII-tubulin, cells were incubated with either 10 nmol/L cabazitaxel or 10 nmol/L docetaxel for 20 h. A 20-h drug incubation period was necessary to insure similar intracellular levels of both drugs since docetaxel is taken up into cells considerably more slowly than cabazitaxel [5]. As shown in Table 2, cabazitaxel more strongly suppressed dynamic instability in control-transfected cells than after βIII-tubulin knockdown. Specifically, after reduction of βIII-tubulin, cabazitaxel inhibited the growing rate by 23% as compared with 29% in control-transfected cells, inhibited the shortening rate by 58 vs. 66%, the shortening length by 70 vs. 82%, and the overall dynamicity by 74% as compared with 85%. In contrast, docetaxel’s suppression of dynamic instability was affected much less by βIII-tubulin knockdown (Table 2). For example, docetaxel’s suppression of the overall dynamicity was similar with or without βIII-tubulin reduction (78% compared with 74%).
We performed two-way ANOVAs to determine whether or not the differences observed between control-transfected and βIII-tubulin-knockdown cells were statistically significant. Data were transformed prior to analysis to meet the assumptions of the ANOVA, and are presented as the back-transformed means in the original scale with error bars showing 95% confidence levels (Fig. 5 and Supplementary Fig. 2; raw data are in Table 2). The results indicated a significant interaction (p < 0.01) between drug treatment (vehicle control, docetaxel, cabazitaxel) and βIII-tubulin content (control-transfected vs. βIII-tubulin-knockdown) for shortening length, shortening rate, and overall dynamicity (Fig. 5a–c). For each of these three parameters, cabazitaxel was significantly less potent in βIII-tubulin-knockdown cells than in control-transfected cells (p < 0.05 for shortening length and rate, and p < 0.01 for overall dynamicity). No such differences were observed for vehicle control or for docetaxel. Collectively, the results indicate that cabazitaxel inhibits dynamic instability in cells more potently when βIII-tubulin is expressed at the normal levels for these cells (~3–5% of total tubulin) than after its reduction by 45%.

Bar graphs show effects of 10 nmol/L cabazitaxel and 10 nmol/L docetaxel on major parameters of microtubule dynamic instability in MCF7 control-transfected cells and after βIII-tubulin knockdown. a–c Show that cabazitaxel’s effects on shortening length, shortening rate, and dynamicity, respectively, are dependent on the status of βIII-tubulin. In contrast, no such differences are observed for vehicle or docetaxel. Data were transformed to meet the assumptions of two-way ANOVA, and all statistical analyses were carried out on the transformed scale. Graphs show mean and 95% confidence levels back-transformed to the original scale. In cases where the two-way ANOVA indicated a significant interaction between drug (veh, cbz, doc) and tubulin type, this is indicated by the p value given at the top of each panel. When a significant interaction was indicated, subsequent pairwise comparisons were carried out using Tukey’s correction for multiple comparisons. Asterisks indicate statistical significance at *p < 0.05, **p < 0.01, ***p < 0.001 levels relative to the appropriate vehicle control, or between conditions as indicated
Discussion
The βIII-isotype of tubulin is unusual [9, 25]. It is predominantly expressed in neurons and in Sertoli cells of the testis and in only small amounts in normal tissues. Of potential clinical significance [15, 26,27,28,29], it is expressed to varying levels in some cancer cells and is highly expressed in aggressive and drug-resistant tumors. The βIII-tubulin isotype has no more than 92% similarity to other β-tubulin isotypes [25]. It contains an unusual distribution of cysteines, can be phosphorylated at a serine near the C-terminus, and, except for the βVI-tubulin isotype, is the only vertebrate β-tubulin isotype that can be phosphorylated in this region [30]. Thus, we investigated its properties and interactions with cabazitaxel, a taxane approved for clinical use [1]. We examined its actions both in vitro with purified bovine brain microtubules containing approximately 25% βIII-tubulin or depleted of βIII-tubulin and in cells with and without a 45% reduction of βIII-tubulin by siRNA.
Examination of the binding of cabazitaxel and docetaxel to purified bovine brain microtubules ± βIII-tubulin (Fig. 1b, c) indicated that the presence or absence of βIII-tubulin made no significant difference in the binding of either drug at concentrations ranging between 0.5 and 40 nmol/L. Cabazitaxel bound to unfractionated microtubules with a K D of 7.4 ± 0.9 (SD) µmol/L and to βIII-depleted microtubules with a K D of 8.3 ± 1.2 (SD) µmol/L; docetaxel’s binding was similar, 6.8 ± 0.2 (SD) µmol/L and 7.9 ± 0.3 (SD) µmol/L, respectively. However, when we examined the effects of 100 nmol/L of each drug on dynamic instability in vitro using microtubules assembled from tubulin containing or devoid of βIII-tubulin and in cells with 10 nmol/L drug after βIII-tubulin knockdown, cabazitaxel clearly inhibited shortening rates, shortening lengths, and overall dynamicity more strongly when βIII-tubulin was present at normal levels than when it was reduced (Tables 1, 2; Figs. 2, 5). In contrast, docetaxel had almost no detectable isotype-dependent effect on these parameters in vitro or in cells (Tables 1, 2; Figs. 2, 5, discussed below).
In MCF7 cells cabazitaxel suppressed dynamic instability more strongly and induced greater levels of mitotic arrest when βIII-tubulin was present at normal levels than after its reduction by siRNA. The differences were modest, but the lack of greater differences may be ascribed to the low quantity of βIII-tubulin in MCF7 cells [31] and to the modest βIII-tubulin knockdown; the level in cells was changed from 3 to 6% βIII-tubulin isotype to 2% βIII-tubulin isotype after knockdown [5]. It is likely that in tumors containing higher levels of βIII-tubulin, as is often the case in drug-resistant aggressive tumors, the enhanced potency of cabazitaxel on βIII-tubulin-rich microtubules may be a significant factor in its efficacy.
These findings raise a puzzling question. If at saturating concentrations the two taxane-based drugs bind with similar affinity to microtubules (Fig. 1b, c) independent of whether βIII-tubulin is present or not, why do they affect microtubule dynamic instability differently in the presence or absence of βIII-tubulin? Molecular dynamics simulations of the binding of paclitaxel, docetaxel, and cabazitaxel to tubulin indicate that cabazitaxel binds substantially differently to tubulin than does either of the other two taxanes, paclitaxel and docetaxel [32]. Therefore, effects of the three drugs on the conformation of the α-β tubulin dimer, its interactions with adjacent tubulin molecules, the stiffness of the protofilaments, and the numbers of microtubule protofilaments vary among the three taxane drugs [32]. Specifically, simulations indicate that upon binding to tubulin cabazitaxel adopts a “collapsed” structure distinctly unlike that of the “T-taxol conformation” adopted upon binding of paclitaxel or docetaxel. Simulations also indicate that cabazitaxel binding to tubulin involves weaker pi-pi interactions, weaker hydrogen bonding, and overall weaker binding than paclitaxel and docetaxel, respectively [32]. Thus, the microtubule binding affinity of various taxanes does not necessarily correlate with their effects on dynamic instability. Discovery of a second taxane binding site within microtubule nanopores also suggests the possibility of yet additional variations between taxanes in their effects on dynamic instability [33].
Another potential source of the differential drug effects we observed with non-saturating taxane concentrations (100 nmol/L in vitro and 10 nmol/L in cells) is provided by the drug binding results (Fig. 1b, c), which indicate that approximately 1 drug molecule (either cabazitaxel or docetaxel) was bound for every 20 tubulin molecules in microtubules at saturating concentrations. Thus, at the sub-saturating concentrations, drug molecules likely bind randomly along a microtubule and would not be crowded or affected by high levels of other drug molecules binding to adjacent tubulin molecules as they are in the saturating conditions used to determine binding affinity. At sub-saturating drug concentrations, the conformational effects of cabazitaxel or docetaxel on adjacent or nearby tubulin molecules may differ depending on the isotype identity of adjacent tubulin molecules, on the random clustering of bound drug, or on isotype-induced altered configurations of adjacent tubulin molecules, thus leading to the possibility of greater suppression of dynamic instability in the presence of βIII-tubulin. Thus, at lower drug concentrations, the stabilizing effects of cabazitaxel or docetaxel on tubulin–tubulin interactions may be affected differently by adjacent tubulin dimers when the microtubule is not saturated with drug.
The principal βIII-tubulin isotype-dependent suppression of microtubule dynamic instability parameters by cabazitaxel was similar both in cells and with purified microtubules. In both, shortening rates, shortening lengths and dynamicity were suppressed less potently by cabazitaxel after βIII-tubulin knockdown (in cells) and after its immunodepletion (in vitro with purified tubulin); compare Table 1 and Fig. 2 (in vitro) with Table 2 and Fig. 5 (in cells). Thus, cabazitaxel suppressed dynamic instability more when βIII-tubulin expression was high (in unfractionated tubulin and in control-transfected cells). In contrast with docetaxel, removal of βIII-tubulin in vitro (Table 1; Fig. 2) or its knockdown in cells (Table 2; Fig. 5) had no appreciable effect on docetaxel suppression of these dynamic instability parameters.
After βIII-tubulin expression siRNA silencing in MCF7 cells, Duran et al, [34] found reduced survival in the presence of cabazitaxel, suggesting that the presence of the βIII-tubulin enhanced resistance to cabazitaxel. However, one issue to be considered in their study is that the degree of knockdown was determined only 2 days after siRNA treatment, whereas survival in the presence of cabazitaxel was tested 14 days after βIII-tubulin knockdown; thus it is not clear what the βIII-tubulin status of the cells was at the time of cell survival determination.
Many studies have examined the effects of βIII-tubulin levels on microtubule-targeted drug actions. It is curious that cabazitaxel appears to be the first example of a taxane-site binding drug that suppresses microtubule dynamics more potently in the presence of βIII-tubulin than after its depletion or reduction. Prior to the observations reported here with cabazitaxel, in general for drugs that bind to tubulin in the paclitaxel binding site (and also in the nearby vinca alkaloid binding site), reduction of βIII-tubulin levels increased drug potency by enhancing suppression of microtubule dynamic instability, whereas increased βIII-tubulin levels were associated with decreased potency and less dynamic instability suppression. Examples include the actions of paclitaxel, estramustine, epothilone B, eribulin, vincristine, and ixabepilone with in vitro polymerized microtubules and in tumor cells [17, 35,36,37,38,39,40]. We note that antagonistic interactions between βIII-tubulin and drug molecules that bind in or near the taxane or vinca sites do not appear to extend to drugs that bind to other sites on tubulin such as peloruside A and laulimalide [41]. Thus, cabazitaxel may be unique among microtubule-targeted drugs identified to date in that it suppresses microtubule dynamic instability more in the presence of βIII-tubulin than after its reduction, a finding that could conceivably lead to development of other drugs with preferred activity on aggressive or drug-resistant cancers associated with expression of high levels of βIII-tubulin.
References
Vrignaud P, Semiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, Riou JF, Commercon A, Lavelle F, Bissery MC (2013) Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res 19(11):2973–2983
Vrignaud P, Semiond D, Benning V, Beys E, Bouchard H, Gupta S (2014) Preclinical profile of cabazitaxel. Drug Des Dev Ther 8:1851–1867
Mitchison TJ, Kirschner M (1984) Dynamic instability of microtubule growth. Nature 312:237–242
Jordan MA, Wilson L (2004) Microtubules as a target for anticancer drugs. Nat Rev Cancer 4(4):253–265
Azarenko O, Smiyun G, Mah J, Wilson L, Jordan MA (2014) Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Mol Cancer Ther 13:2092–2103
Field JJ, Kanakkanthara A, Miller JH (2014) Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem 22(18):5050–5059
Kamath K, Smiyun G, Wilson L, Jordan MA (2014) Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskelet (Hoboken) 71(1):46–60
Lu Q, Luduena RL (1994) In vitro analysis of microtubule assembly of isotypically pure tubulin dimers. Intrinsic differences in the assembly properties of alpha beta II, alpha beta III, and alpha beta IV tubulin dimers in the absence of microtubule-associated proteins. J Biol Chem 269(3):2041–2047
Ludueña RF, Banerjee A (2008) The isotypes of tubulin. In: Fojo AT (ed) The role of microtubules in cell biology, neurobiology and oncology, 1st edn. Humana Press, Totowa, NJ, pp 123–175
Kavallaris M, Kuo DY-S, Burkhardt CA, Regl DL, Norris MD, Haber M, Horwitz SB (1997) Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest 100(5):1–12
Gan PP, Pasquier E, Kavallaris M (2007) Class III beta-tubulin mediates sensitivity to chemotherapeutic drugs in non small cell lung cancer. Cancer Res 67(19):9356–9363
Stengel C, Newman SP, Leese MP, Potter BV, Reed MJ, Purohit A (2010) Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br J Cancer 102(2):316–324
Ploussard G, Terry S, Maille P, Allory Y, Sirab N, Kheuang L, Soyeux P, Nicolaiew N, Coppolani E, Paule B, Salomon L, Culine S, Buttyan R, Vacherot F, de la Taille A (2010) Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res 70(22):9253–9264
McCarroll JA, Gan PP, Liu M, Kavallaris M (2010) BetaIII-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer Res 70(12):4995–5003
McCarroll JA, Sharbeen G, Liu J, Youkhana J, Goldstein D, McCarthy N, Limbri LF, Dischl D, Ceyhan GO, Erkan M, Johns AL, Biankin AV, Kavallaris M, Phillips PA (2015) betaIII-tubulin: a novel mediator of chemoresistance and metastases in pancreatic cancer. Oncotarget 6(4):2235–2249
Miller HP, Wilson L (2010) Preparation of microtubule protein and purified tubulin from bovine brain by cycles of assembly and disassembly and phosphocellulose chromatography. Methods Cell Biol 95:3–15
Lopus M, Smiyun G, Miller H, Oroudjev E, Wilson L, Jordan MA (2015) Mechanism of action of ixabepilone and its interactions with the betaIII-tubulin isotype. Cancer Chemother Pharmacol 76(5):1013–1024
Yenjerla M, Lopus M, Wilson L (2010) Analysis of dynamic instability of steady-state microtubules in vitro by video-enhanced differential interference contrast microscopy with an appendix by Emin Oroudjev. Methods Cell Biol 95:189–206
R Core Team (2016) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Fox J, Weisberg S (2011) An R companion to applied regression, 2nd edn. Sage, Thousand Oaks
RStudio Team (2016) RStudio: Integrated development for R. RStudio Team, Boston
Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer, New York
Panda D, Miller HP, Banerjee A, Luduena RF, Wilson L (1994) Microtubule dynamics in-vitro are regulated by the tubulin isotype composition. Proc Natl Acad Sci USA 91(24):11358–11362
Hiser L, Aggarwal A, Young R, Frankfurter A, Spano A, Correia JJ, Lobert S (2006) Comparison of beta-tubulin mRNA and protein levels in 12 human cancer cell lines. Cell Motil Cytoskelet 63(1):41–52
Luduena RF (1998) Multiple forms of tubulin: different gene products and covalent modifications. Int Rev Cytol 178:207–275
Kavallaris M, Kuo DY-S, Burkhardt CA, Regl DL, Norris MD, Haber M, Horwitz SB (1997) Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest 100:1–12
Gan PP, Pasquier E, Kavallaris M (2007) Class III beta-tubulin mediates sensitivity to chemotherapeutic drugs in non small cell lung cancer. Cancer Res 67(19):9356–9363
Ploussard G, Terry S, Maille P, Allory Y, Sirab N, Kheuang L, Soyeux P, Nicolaiew N, Coppolani E, Paule B, Salomon L, Culine S, Buttyan R, Vacherot F, de la Taille A (2010) Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res 70(22):9253–9264
McCarroll JA, Gan PP, Liu M, Kavallaris M (2010) BetaIII-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer Res 70(12):4995–5003
Khan I, Luduena R (1996) Phosphorylation of betaIII-tubulin. Biochemistry 35:3704–3711
Lobert S, Jefferson B, Morris K (2011) Regulation of beta-tubulin isotypes by micro-RNA 100 in MCF7 breast cancer cells. Cytoskelet (Hoboken) 68(6):355–362
Churchill CD, Klobukowski M, Tuszynski JA (2015) Elucidating the mechanism of action of the clinically approved taxanes: a comprehensive comparison of local and allosteric effects. Chem Biol Drug Des 86(5):1253–1266
Freedman H, Huzil JT, Luchko T, Luduena RF, Tuszynski JA (2009) Identification and characterization of an intermediate taxol binding site within microtubule nanopores and a mechanism for tubulin isotype binding selectivity. J Chem Inf Model 49(2):424–436
Duran GE, Wang YC, Francisco EB, Rose JC, Martinez FJ, Coller J, Brassard D, Vrignaud P, Sikic BI (2015) Mechanisms of resistance to cabazitaxel. Mol Cancer Ther 14(1):193–201
Derry WB, Wilson L, Khan IA, Luduena RF, Jordan MA (1997) Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry 36(12):3554–3562
Gan PP, McCarroll JA, Po’uha ST, Kamath K, Jordan MA, Kavallaris M (2010) Microtubule dynamics, mitotic arrest, and apoptosis: drug-induced differential effects of betaIII-tubulin. Mol Cancer Ther 9(5):1339–1348
Kamath K, Wilson L, Cabral F, Jordan MA (2005) BetaIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J Biol Chem 280(13):12902–12907
Laing N, Dahllof B, Hartley-Asp B, Ranganathan S, Tew KD (1997) Interaction of estramustine with tubulin isotypes. Biochemistry 36(4):871–878
Narvi E, Jaakkola K, Winsel S, Oetken-Lindholm C, Halonen P, Kallio L, Kallio MJ (2013) Altered TUBB3 expression contributes to the epothilone response of mitotic cells. Br J Cancer 108(1):82–90
Wilson L, Lopus M, Miller HP, Azarenko O, Riffle S, Smith JA, Jordan MA (2015) Effects of eribulin on microtubule binding and dynamic instability are strengthened in the absence of the betaIII tubulin isotype. Biochemistry 54(42):6482–6489
Kanakkanthara A, Eras J, Northcote PT, Cabral F, Miller JH (2014) Resistance to peloruside A and laulimalide: functional significance of acquired betaI-tubulin mutations at sites important for drug-tubulin binding. Curr Cancer Drug Targets 14(1):79–90
Acknowledgements
This work was supported by a Grant from Sanofi-Aventis, Cambridge (MA), and the NRI-MCDB Microscopy Facility at UC Santa Barbara (NIH #1 S10 OD010610-01A1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sanofi-Aventis (Grant Number CW70839).
Conflict of interest
Author Mary Ann Jordan has received a research grant from Sanofi-Aventis and has received a speaker honorarium, but does not own any stock in Sanofi-Aventis. Author Smiyun declares that he has no conflict of interest, author Azarenko declares that she has no conflict of interest, Author Miller declares that he has no conflict of interest, Author Rifkind declares that he has no conflict of interest, Author LaPointe declares that she has no conflict of interest, and Author Wilson declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
280_2017_3345_MOESM2_ESM.eps
Supplementary Fig. 1 Effects of vehicle (veh), 100 nmol/L cabazitaxel (cbz) and 100 nmol/L docetaxel (doc) on major parameters of dynamic instability in microtubules assembled in vitro with unfractionated bovine brain tubulin (“Unfractionated”) or with bovine brain tubulin depleted of βIII-tubulin (“βIII-depleted”). Panels show (A) growth length, (B) growth rate, (C) catastrophe frequency, and (D) rescue frequency. The left half of each panel is a bar graph of the raw mean ± SEM or back-transformed means with error bars showing 95% confidence levels, as indicated on the y-axis, and the right half of each panel shows density plots of the raw data. Each panel indicates whether data were analyzed by two-way ANOVA or by the non-parametric Kruskal–Wallis test, and significant results of any post hoc tests are shown. Asterisks indicate statistical significance at *p < 0.05, **p < 0.01, and ***p < 0.001 relative to the appropriate vehicle control or as indicated (EPS 3849 kb)
280_2017_3345_MOESM3_ESM.eps
Supplementary Fig. 2 Effects of 100 nmol/L cabazitaxel (cbz) and 100 nmol/L docetaxel (doc) on major parameters of dynamic instability in microtubules in MCF7 control-transfected cells and in MCF7 cells after βIII-tubulin knockdown. Panels show (A) growth length, (B) growth rate, (C) catastrophe frequency, and (D) rescue frequency. The left half of each panel is a bar graph of the raw means +/− SEM or back-transformed means with error bars showing 95% confidence levels, as indicated on the y-axis, and the right half of each panel shows density plots of the raw data. Each panel indicates whether data were analyzed by two-way ANOVA or by the non-parametric Kruskal–Wallis test, and significant results of any post hoc tests are shown. Asterisks indicate statistical significance at *p < 0.05, **p < 0.01, and ***p < 0.001 relative to the appropriate vehicle control or as indicated (EPS 6017 kb)
Rights and permissions
About this article

Cite this article
Smiyun, G., Azarenko, O., Miller, H. et al. βIII-tubulin enhances efficacy of cabazitaxel as compared with docetaxel. Cancer Chemother Pharmacol 80, 151–164 (2017). https://doi.org/10.1007/s00280-017-3345-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-017-3345-2