
Abstract
The mechanistic pathways for the sequential tandem [4 + 2] / [3 + 2] versus [3 + 2] / [4 + 2] cycloaddition reaction of functionalized acetylenes with cyclopentadiene and dimethyl diazopropane for the formation of norbornene pyrazolines, employed in the synthesis of pharmaceutically relevant compounds, have been studied computationally with DFT at the M06-2X/6-31G(d) and M06-2X/6-31G(d,p) levels of theory. We have established that, in the reaction of the parent (unsubstituted) acetylene with cyclopentadiene and dimethyl diazopropane, the order of the tandem addition has no substantial effects in product outcomes. The same product is obtained provided the reaction components remain the same for both [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] tandem addition sequences. The results indicate that the [4 + 2] Diels–Alder addition step is the rate-determining step irrespective of the addition order, while the 1,3-dipolar cycloaddition step has been found to generally proceed rapidly with very low activation energies. It has also been realized that the regio-, stereo-, and chemo-selectivities of the reaction are strictly dictated by the type of substituent on the parent acetylene. For substituted acetylenes, we conclude that the sequence of the tandem addition generally affects the type of isomeric product obtained. The [4 + 2]/[3 + 2] tandem addition sequence has been established to favor the exo stereo-selective isomer over the endo, whereas the [3 + 2]/[4 + 2] tandem addition sequence generally favors the endo product formation. Therefore, it is settled that the mechanistic route taken by any substrate in the [4 + 2]/[3 + 2] versus [3 + 2]/[4 + 2] sequential tandem cycloaddition is greatly affected by both electronic and steric factors. Electrophilicity indices calculations agree with the activation barriers obtained. Perturbation molecular orbital theory was employed to rationalize the results. Global reactivity indices calculations gave a good correlation with the activation energies.

The sequential tandem [4 + 2] / [3 + 2] versus [3 + 2] / [4 + 2] cycloaddition reaction of functionalized acetylenes with cyclopentadiene and dimethyl diazopropane for the formation of norbornene pyrazolines.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The efficient construction of complex molecules from simpler ones is among the major objectives in organic syntheses. Consequently, multiple chemical transformations in a single conversion are a crucial for high synthetic efficiency. Tandem reaction is one of the renowned synthetic strategies for the rapid construction of complex structures. Pericyclic reaction is a well-known approach in the domain of tandem reactions. Pericyclic reactions comprising at least one cycloaddition have attracted widespread application in organic synthesis [1,2,3].
A review by Denmark and coworker [2] categorized tandem cycloaddition reactions into three, comprising tandem cascade, consecutive, and sequential cycloaddition reactions. In tandem cycloadditions, the reaction is initiated with two reactive components to yield an intermediate. In some instances, the intermediate may be stable enough for isolation. The third reactive component is introduced in a separate step upon generation of the required functionalities in the intermediate for the reaction to proceed to finality [4]. The work of Danishefsky and his team [5], as shown in (Scheme 1), clearly exemplifies this class of reaction in the syntheses of vernolepin and vernomenin.

Syntheses of vernolepin and vernomenin by Danishefsky et al. [5]
Neumann and his coworkers [6] reported a tandem [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition sequence by reacting cyclopentadiene with a range of functionalized acetylenes and a diazopropane as shown in Scheme 2. In their work, they treated dimethyl acetylenedicarboxylate with cyclopentadiene in a [4 + 2] addition fashion, which yielded the appropriate cycloadduct. Upon a subsequent reaction of the Diels–Alder adduct with a third reactive component (2-diazoalkane), it furnished them the stereoselective norbornene pyrazoline and its derivatives [6]. The tandem sequential [4 + 2]/[3 + 2] cycloadduct was isolated as a single diastereomer as depicted in Scheme 2 above. Based on the stereochemistry of the tandem adduct obtained, Neumann and his team reasoned that the final product of the [4 + 2]/[3 + 2] tandem sequential addition sequence/order resulted from an exo attack by the 1,3-dipole to the dipolarophile. However, a sharp discovery was made that reversing the reaction order from [4 + 2]/[3 + 2] to [3 + 2]/[4 + 2] yielded a stereoisomer of the same tandem adduct as the major product as shown in Scheme 2.

Neumann and his coworkers [6] tandem [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition of cyclopentadiene with dimethyl acetylenedicarboxylate and a diazopropane
The utility of the tandem [4 + 2]/[3 + 2] of any sequence has been extensively employed by Denmark and his group in the construction of complex diverse heterocycles [7,8,9] as well as other researchers [10]. The usefulness of this method can be revealed in its extensive applications in synthetic organic chemistry for the construction of various heterocyclic compounds [11,12,13,14,15,16].
In recent times, in view of the useful applications of pyrazoline and its derivatives, several studies have been devoted towards their syntheses. Pyrazoline and its derivatives, the products of the tandem [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition of cyclopentadiene with functionalized alkynes and a diazopropane, have been extensively reported to possess several biological activities [10, 17,18,19].
Despite all the progress, the only method available for the synthesis of norbornene pyrazoline derivatives is the Neumann’s tandem [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] addition sequences by reacting cyclopentadiene with a range of functionalized acetylenes and a diazopropane. In this reaction, several regio-, stereo- and chemo-selective tandem adducts are possible to obtain. However, there is no mechanistic study on the tandem sequential [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition reaction of cyclopentadienes with acetylenes and diazoalkanes to establish the reactivity and the origin of the regio-, stereo-, and chemo-selectivities of the reaction. In addition, no experimental nor theoretical study has been conducted on the substitution effects on the tandem Diels–Alder / 1, 3 – dipolar cycloaddition of cyclopentadienes with acetylenes and diazoalkanes pathways of any order to ascertain how it may affect selectivity and efficiency. These mechanistic considerations are very crucial towards the syntheses of norbornene pyrazolines of high selectivity and efficiency for improved activity.
Herein, we employ density functional theory (DFT) calculations to elucidate the mechanism of the reaction of the modeled cyclopentadiene with a range of acetylenes and diazopropane (Scheme 3), with the aim of shedding light on the molecular level mechanistic details of the tandem sequential [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition reaction of cyclopentadienes with acetylenes and diazopropanes. This study also investigates the effects of substituents on the reaction pathways as well as regio-, stereo-, and chemo-selectivities of the reaction as spelt out in Scheme 3.

Proposed reaction pathways for the study [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] cycloaddition reaction of cyclopentadienes with acetylenes and diazopropanes
Computational details and methodology
We performed the DFT computations using the Spartan’14 [20] and Gaussian 09 [21] Molecular Modeling software packages at the M06-2X level of theory with the 6-31G(d) and 6-31G(d,p) basis sets. The M06-2X functional [22] is a hybrid meta-generalized gradient approximation (meta-GGA) density functional, and has been found to be effective at computing thermochemical and kinetic parameters, for reactions where nonlocal dispersion interactions play a dominant role [16, 23,24,25,26]. Houk and coworkers have shown that M06-2X generally averts systematic errors in energetic barrier heights and reaction energies present with, for instance, B3LYP in reactions involving predominant changes in C-C bonding [27, 28]. Houk and his team have compared energetics at the relatively modest 6-31G(d) basis set with single-point calculations with a larger 6-311G(d,p) basis set and established that the larger basis set size does not affect the conclusions [26].
The starting geometries of the molecular systems were built using Spartan’s graphical model builder and minimized interactively using the SYBYL force field [29]. We computed transition state structures by first obtaining guess input structures. This was done by constraining specific internal coordinates of the molecules (bond lengths, bond angles, dihedral angles) while fully optimizing the remaining internal coordinates. This procedure gives appropriate guess transition state input structures, which are then submitted for full transition state calculations without any geometry or symmetry constraints. The full optimization of all structures was carried out with the Gaussian 09 computational chemistry software package. Full vibrational frequency calculations verified that each transition state structure had a Hessian matrix with only a single negative eigen value, characterized by an imaginary vibrational frequency along the respective reaction coordinate. Intrinsic reaction coordinate calculations were then performed to ensure that each transition state smoothly connects the reactants and products along the reaction coordinate [30, 31]. Using benzene, the polarizable continuum model (PCM) was employed to model the effects of solvents in the reactions [32].
The global electrophilicities (ω) and maximum electronic charge (ΔNmax) of the various acetylene derivatives were calculated using Eqs. (1) and (2). The electrophilicity index has been used as a parameter for the analysis of the chemical reactivity of molecules. It measures the ability of a reaction substrate to accept electrons [33] and is a function of the electronic chemical potential, μ = (EHOMO + ELUMO)/2 and chemical hardness, η = (ELUMO - EHOMO) as defined by Pearson’s acid-base concept [34]. Species with large electrophilicity values are more reactive towards nucleophiles. These equations are based on the Koopmans’ theorem [35] originally established for calculating ionization energies from closed-shell Hartree–Fock wavefunctions, but have since been adopted as acceptable approximations for computing electronic chemical potential and chemical hardness.
The maximum electronic charge transfer (ΔNmax) measures the maximum electronic charge that the electrophile may accept. Thus, species with large ΔNmax index would be best electrophile given a series of compounds.
Results and discussion
[4 + 2] / [3 + 2] sequential tandem addition reaction of the parent unsubstituted acetylene with cyclopentadiene and diazopropane
Scheme 3 shows the proposed pathways for the reaction of functionalized acetylenes with cyclopentadiene and 2-diazopropane. In the Diels–Alder / 1,3-dipolar addition sequence, the reaction of the functionalized acetylenes (A) and cyclopentadiene (B) affords the corresponding Diels–Alder adduct 4Int through the transition state 4TS1. The cycloadduct intermediate undergoes subsequent 1,3-dipolar cycloaddition reaction with the 2-diazopropane (C). In order to ascertain the regioselectivity of the reaction, we proposed the addition of the 1,3-dipole to the dipolarophile via two possible routes, i.e., across the substituted and unsubstituted olefinic bonds in the in situ-generated dipolarophile intermediate. The addition of the 2-diazopropane across the substituted olefinic bond in the norbornadiene can occur through endo or exo transition states, 4TS2AEndo or 4TS2AExo to yield either 4PdtAEndo or 4PdtAExo. On the possibility that the addition occurs across the unsubstituted double bond in the norbornadiene, it can do so through the endo or exo transition states 4TS2BEndo or 4TS2BExo to form the corresponding 4PdtBEndo or 4PdtBExo, respectively.
Changing the sequence of the reaction (but maintaining the same components and reaction conditions), we envisage that the concerted [3 + 2] addition of the 2-diazopropane (C) to the functionalized-acetylene (A) to yield the cycloadduct 3Int through transition states 3TS1. The subsequent [4 + 2] addition of the resulting cycloadduct intermediate 3Int to the cyclopentadiene (B) can occur through the exo and endo transition states 3TS2AExo and 3TS2AEndo to yield the corresponding PdtAExo or PdtAEndo, respectively. We also explored the chemoselectivity of the addition of the cyclopentadiene (B) across the N=N linkage in the pyrazole cycloadduct intermediate 3Int through transition state TS2N=N to furnish PdtN=N.
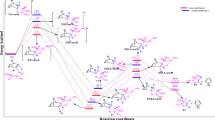
Figure 1 shows the optimized geometries of the stationary points (minima and maxima) relevant to the proposed scheme of study as well as the zero-point-corrected Gibbs free-energy profile of the [4 + 2]/[3 + 2] addition of parent (unsubstituted) acetylene with pyropentylene and 2-diazopropane. From the profile, the activation barrier through the 4TS1 transition state is 19.4 kcal/mol, making it the rate-determining step along the [4 + 2]/[3 + 2] addition sequence pathway. Although the transition state 4TS1 leads to the formation of a very stable intermediate 4Int with energy of − 33.2 kcal/mol, the subsequent addition to the 2-diazopropane (C) requires only an activation barrier of 10.0 kcal/mol and 10.2 kcal/mol for the exo and endo stereo-selective isomers, respectively. This implies that the energetic difference between the 4TS2AExo and the 4TS2AEndo is only 0.2 kcal/mol, suggesting very competitive routes to the formation of the final products. Hence, the stability of the final products (4PdtAExo and 4PdtAEndo) becomes crucial as to which one is observed. The exo product (4PdtAExo) is marginally more stable than the competitive endo isomeric product (4PdtAEndo) by 1.9 kcal/mol exergonic. Considering the fact that the products are so stable (− 86.1 kcal/mol and − 84.2 kcal/mol for the 4PdtAExo and 4PdtAEndo, respectively, the possibility of the reaction being reversible is limited. Hence, it can be said that the reaction is strictly controlled by kinetic factors, which as the barriers show, point to the formation of both the exo and endo products. The reported energies are relative to the starting materials. The relevant geometrical parameters such as bond lengths associated with structural changes as the reaction proceeds from reacts to the tandem adducts are shown in Fig. 1.

Zero-point energy-corrected Gibbs free-energy profile of the [4 + 2]/[3 + 2] addition of unsubstituted acetylene with cyclopentadiene and 2-diazopropane at the M06-2X with the 6-31G(d) basis set. Relative energies in kcal mol−1. All bond distances are measured in Å
[3 + 2] / [4 + 2] sequential tandem addition reaction of the parent unsubstituted acetylene with cyclopentadiene and diazopropane
The optimized geometries of the reactants, intermediates, transitions states, as well as the products involved in the 1,3 – dipolar [3 + 2] addition of the unsubstituted acetylene and the 2-diazopropane followed by the Diels–Alder [4 + 2] addition of the cycloadduct intermediate and the cyclopentadiene are shown on Fig. 2.

Zero-point-corrected Gibbs free-energy profile of the [3 + 2]/[4 + 2] addition of unsubstituted acetylene with 2-diazopropane and cyclopentadiene at the M06-2X with the 6-31G(d) basis set. Relative energies in kcal mol−1. All bond distances are measured in Å
Here, the initial [3 + 2] dipolar cycloaddition between the 2-diazopropane and the unsubstituted acetylene occurs via a transition state 3TS1 with an activation barrier of 11.6 kcal/mol. Transition state 3TS1 leads to the formation of a very stable intermediate 3Int, with a reaction energy of − 57.8 kcal/mol. It is worth noting that by changing the addition sequence to the [3 + 2]/[4 + 2], the rate-determining step changes from the first activation barrier to the second activation energy barrier. This is an indication that the [3 + 2] dipolar cycloaddition of these reaction components may generally occur more rapidly than the [4 + 2] Diels–Alder addition reaction. The Diels–Alder reaction between the in situ-generated pyrazole (3Int) and the cyclopentadiene proceeds via the exo and endo transition states 3TS2AExo and 3TS2AEndo towards the formation of the isomeric products 3PdtExo and 3PdtEndo. We found the activation barrier for the exo isomer to be 15.0 kcal/mol while that of the endo isomer is 15.2 kcal/mol. This signifies an energetic difference of only 0.1 kcal/mol, suggesting very competitive routes. Therefore, the stability of the corresponding final products become very critical in determining which products are obtained as aforementioned in the case of the [4 + 2]/[3 + 2] addition sequence. Results from our calculations show that the reaction energy of the exo isomer is − 86.1 kcal/mol while that of the endo isomer is − 84.2 kcal/mol. Therefore, owing to the observation that both products are very stable, the reaction will most likely be controlled solely by kinetic factors competitively in favor of both the exo and endo isomeric products. Our proposed chemoselective addition of the cyclopentadiene across the N=N linkage in the pyrazole intermediate was found to be an unfavorable pathway compared to its corresponding exo and endo diastereomers. The transition state along this pathway, TS2N=N was found to have an activation height of 20.6 kcal/mol. This indicates that this pathway is ≈ 5.0 kcal/mol higher than the competitive endo (3TS2AEndo) and exo (3TS2AExo) isomeric pathways, making it a disfavored pathway. Again, we found its corresponding product, PdtN=N, to have a reaction energy of 62.0 kcal/mol exergonic, being the least stable among the three proposed products along this [3 + 2]/[4 + 2] addition pathway.
[4 + 2] / [3 + 2] sequential tandem addition reaction of dimethyl but-2-ynedioate with cyclopentadiene and dimethyl diazopropane
Based on the results obtained from the reaction of the unsubstituted acetylene with the other respective components, we extend the studies to substituted acetylenes. Here, we explore the [4 + 2] / [3 + 2] addition of dimethyl acetylenedicarboxylate with cyclopentadiene and 2-diazopropane. This part of the work employs the same reaction components used by Newman and coworkers [6] in their experimental studies. The optimized structures of all the reactants, intermediates, products, and transition states as well as their relative energetics and geometrical parameters are shown in Figs. 3 and 4. We found the Diels–Alder [4 + 2] addition step to proceed through transition state 4TS1-Ester with an activation barrier of 10.4 kcal/mol. This step was found to be the rate-determining step. The transition state 4TS1- Ester leads to formation of a very stable intermediate 4Int-Ester, which is 43.4 kcal/mol more stable than the reactants. However, the subsequent [3 + 2] addition with the 1,3-dipole requires a very small activation barrier of 0.2 kcal/mol and 1.6 kcal/mol along the exo and endo stereoselective isomers across the substituted olefinic bond in the norbornadiene, a further indication that the 1,3-dipolar cycloaddition occurs very rapidly. Considering the addition of the diazoalkane across the unsubstituted olefinic bond in the norbornadiene, we obtained an activation energy of 10.3 kcal/mol for the exo isomer (TS2BExo-Ester) and 10.4 kcal/mol for the endo isomer (TS2BEndo-Ester). Based on these results, it can said that the addition of the 2-diazopropane to the norbornadiene will occur competitively between the endo and exo isomers across the ester-substituted olefinic bond of the dipolarophile. The corresponding stereoisomeric products 4PdtAExo-Ester and the 4PdtAEndo-Ester have reaction energies of − 95.3 kcal/mol and − 92.0 kcal/mol, respectively. The observed energetic trends account for why Newman and his coworkers obtained only the exo (4PdtAExo-Ester) isomer of the tandem adduct. The 3D representations and the relevant bond distances of all the structures are reported in Fig. 4.

Zero-point energy-corrected Gibbs free-energy profile of the [4 + 2]/[3 + 2] addition of dimethyl but-2-ynedioate with cyclopentadiene and 2-diazopropane at the M06-2X with the 6-31G(d) basis set. Relative energies in kcal mol−1. All bond distances are measured in Å

Optimized geometries of the transitions states and equilibrium geometries involved in the tandem sequential [4 + 2]/[3 + 2] addition of dimethyl but-2-ynedioate with dimethyl diazopropane and cyclopentadiene at the M06-2X with the 6-31G(d) basis set. All bond distances are measured in Å. Atomic color code (red = oxygen, blue = nitrogen, gray = carbon)
[3 + 2]/[4 + 2] sequential tandem addition reaction of dimethyl but-2-ynedioate with cyclopentadiene and dimethyl diazopropane
We explored the potential energy surface of the reaction of dimethyl acetylenedicarboxylate with cyclopentadiene and the dimethyl diazopropane via the [3 + 2]/[4 + 2] addition sequence. Figures 5 and 6 present the optimized structures, geometrical parameters, as well as the zero-point energy-corrected Gibbs free activation and reaction energies along the [3 + 2]/[4 + 2] addition pathways for the sequential tandem addition reaction of dimethyl but-2-ynedioate with cyclopentadiene and dimethyl diazopropane. The reaction of the diester-substituted acetylene and the dimethyl diazopropane via [3 + 2] addition fashion occurs through a rapidly occurring transition state 3TS1-Ester with an energy barrier of a just 0.6 kcal/mol. Transition state 3TS1-Ester leads to the formation of a very stable pyrazole intermediate, 3Int-Ester. Subsequent reaction of the pyrazole intermediate to the diene via a [4 + 2] Diels–Alder addition fashion through stereoisomeric transition states 3TS2AExo-Ester and 3TS2AEndo-Ester with activation energies of 8.6 kcal/mol and 11.0 kcal/mol, respectively. Thus, we found that the 3TS2AExo-Ester is lower than the 3TS2AEndo-Ester by 2.4 kcal/mol. The corresponding stereoisomeric products 3PdtAExo-Ester and the 3PdtAEndo-Ester have reaction energies of 93.9 kcal/mol and 90.5 kcal/mol, respectively. Therefore, the exo product is more stable than the endo isomeric product. Hence, the exo pathway is favored for both thermodynamic and kinetically controlled reaction although with marginal energy differences. From scheme 2, it can be seen that the yields of the reactions are in concordance with the energy analyses.

Zero-point energy-corrected Gibbs free-energy profile of the [3 + 2]/[4 + 2] addition of dimethyl but-2-ynedioate with dimethyl diazopropane and cyclopentadiene at the M06-2X with the 6-31G(d) basis set. Relative energies in kcal mol−1. All bond distances are measured in Å

Optimized geometries of the transitions states and equilibrium geometries involved in the tandem sequential [3 + 2]/[4 + 2] addition of dimethyl diazopropane with cyclopentadiene and dimethyl but-2-ynedioate at the M06-2X with the 6-31G(d) basis set. All bond distances are measured in Å. Atomic color code (red = oxygen, blue = nitrogen, gray = carbon)
[4 + 2]/[3 + 2] sequential tandem cycloaddition addition reaction of substituted acetylenes with cyclopentadiene and dimethyl diazopropane
To investigate the effects of substituents on the energetic trends and the isomeric selectivity of the reaction, we introduce varying substituents at position R on the acetylene (Scheme 3). All the results of the substituents considered in this work are presented on Tables 1 and 2. Starting from electron-donating groups, we introduced a dimethyl group on the acetylene to obtain A(CH3)2. The reaction of the 2-butyne occurred through transition state 4TS1-CH3 with an activation energy of 22.6 kcal/mol, which is 3.4 kcal/mol higher than the parent acetylene. Although the first transition state (rate-determining step) along this path is higher than the parent acetylene, we found its corresponding product to be less stable than the parent by 3.6 kcal/mol. For the formation of the final products through all of the four proposed transition states, we found 4TS2BExo-CH3 to be the most favored. This observation is in sharp contrast to that seen for the parent acetylene and dimethyl acetylenedicarboxylate as discussed earlier. We also found the corresponding product for the 4TS2BExo-CH3 to be the most stable with a reaction energy of − 83.0 kcal/mol. Therefore the reaction of 2-butyne with the respective components favor regioselective addition across the unsubstituted olefinic bond of the dipolarophile, which is contrary to the observed product in the parent acetylene.
When we introduced methoxy groups on the acetylene to from A(OCH3), we observed analogous energetic trends as that of the 2-butyne, although we found a reduction in the rate-determining transition state (4TS1-Methoxy) by 2.5 kcal/mol. Here again, the results show that addition across the unsubstituted olefinic bond in the norbornadiene intermediate is highly favored over all the other region- and stereo-isomeric transition states. We also realized that its corresponding product (4PdtBExo-Methoxy) is the most stable among the four proposed products.
All other electron-releasing groups considered in this work (OH, NH2) favored the addition of the dipole 2-diazopropane) across the unsubstituted olefinic bond in the dipolarophile. Therefore, it can be said that the addition of electron-donating substituted alkynes to the cyclopentadiene and the diazoalkane promotes both regio and stereo-selectivities towards the formation of the 4PdtBExo products.
We also extended the scope of the study to electron-withdrawing substituted acetylenes (Scheme 3). For all the electron-withdrawing groups considered in the [4 + 2] / [3 + 2] addition, we observed a drastic decrease in the first transition states (rate-determining step). For diamide substituted acetylene, the [4 + 2] addition step proceeds with an activation barrier of 11.0 kcal/mol leading to the formation of the 4Int with reaction energy of − 4.5. We found 4TS2AExo-Amide, which occurs across the substituted olefinic bond in the dipolarophile, to be the most favored with an activation height of 5.1 kcal/mol. We also observed its corresponding product to be much more stable than its endo isomeric product. Also, for CN and CF3 substituted acetylene to form A(CN)2 and A(CF3)2, respectively, we observed similar energetic trends. In all cases, we found a drastic reduction in the barrier height of the rate-determining step. The subsequent [3 + 2] addition reaction also favors the 4TS2AExo pathway in both cases. However, for a Cl-substituted acetylene to yield A(Cl)2, we noticed a strong reduction in the rate-determining activation barrier. We also established the second step of the reaction ([3 + 2] addition step) to proceed in favor of the direct regio-isomer observed for all the electron-accepting groups on the parent acetylene. Its corresponding product is found to be the most favored, as shown in Table 1. We also explored the nature of the potential energy surface when cyclopropane is introduced on the acetylene at position R (Scheme 3). We realized an increase in activation energy of the rate-determining step leading to the formation of relatively less stable pyrazole intermediate compared to the parent acetylene. Interestingly, for all the four isomeric transition states along the [3 + 2] addition step, we found the 4TS2AExo-Cyclopropane to be the most favored. We also realized its corresponding product 4PdtBExo-Cyclopropane to be the most favored. This observation agrees with that observed in the case of electron-donating substituted acetylenes. This implies that the reaction is both kinetically and thermodynamically favored along the 4TS2BExo-Cyclopropane pathway leading to the formation of the corresponding product, 4PdtExo-Cyclopropane.
[3 + 2]/[4 + 2] sequential tandem cycloaddition addition reaction of substituted acetylenes with cyclopentadiene and dimethyl diazopropane
In order to investigate the effects of substituents on the acetylene along the [3 + 2]/[4 + 2] addition reaction pathways, we employed various electron-withdrawing, electron-donating, and ring structures on the parent acetylene, as shown in Table 2. For all the electron-donating groups considered in this section, we saw a general activation energy increase in the [4 + 2] addition step, which is the rate-determining step. For dimethoxy (OCH3)2, and dihydroxyl (OH)2, substituted acetylene, the results show that the stereo-selective endo pathway is favored.
Formation of the endo products in the reaction of A(OCH3)2 and A(OH)2 are preferred by kinetic factors. However, the reaction of the diamino acetylene A(NH2)2 favors exo selective isomer although attempts to locate TS2N=N-Amino was unsuccessful. We observed 22.7 kcal/mol and 23.5 kcal/mol energy barriers, respectively, for the 3TS2AExo-Amino and 3TS2AEndo-Amino. In addition, we found the PdtN=N-Amino to be the least stable for the three proposed products along this pathway.
We further made an exploration on how introducing electron-accepting groups on the parent acetylene will affect reactivity and selectivity of the reaction. It was discovered that for diamide acetylene A(CONH2)2 and dichloro acetylene A(Cl)2, the endo pathway is favored over the exo and the chemo-selective pathways, although the corresponding products in each case are less stable than the exo tandem adduct. In the case of dicyano, A(CN)2 and A(CF3)2, the exo pathway was found to be formed over the endo and N=N routes. Again, their corresponding products are also found to be more stable than the endo and N=N products. Finally, we introduced dicyclopropene on the acetylene. It was realized that the formation of the exo tandem adduct is favored under thermodynamic considerations.
From Tables 1 and 2, it can be seen that some of the activation barriers were slightly negative. We reasoned that this could be due to basis set superposition error. In order to ascertain this reason, we employed the 6-31G(d,p) basis set in some selected substrates. Indeed, results from Tables 3 and 4 indicate an appreciable increase in activation barriers among substrates where slightly negative barriers were recorded earlier with the relatively modest 6-31G(d) basis set. It should be noted that when the basis set was changed to 6-31G(d,p), there was an appreciation of the energetics with no negative activation barrier observed. However, the energetic trends remained the same as that observed when the relatively modest 6-31G(d) was used.
Global electrophilicities and maximum electronic charges
We extended the scope of this work by calculating the electrophilicity indices (ω) and maximum electronic charge transfer (ΔNmax) of the various acetylene derivatives considered in this work. As alluded to, substrates with large ω values are more reactive towards nucleophiles. Indeed, trends in calculated ω values are in agreement with the DFT-calculated activation energies of the reaction presented in the previous sections. For instance, we observed that CH3 and NH2 substrates had the least ω values, making them the least reactive substrates, which accounts for the highest activation barriers for these chemical species. Analogous observations were also made for the ΔNmax values, where substrates with small ΔNmax values were found to have low energy barriers as reported in Table 5. When the activation energies are plotted versus the computed electrophilicity ω indices of the various acetylene derivatives, a good linear correlation is obtained (R2 = 0.76; see Fig. 7).

Plot of the M06-2X/6-31G(d) computed first activation energy barriers along the [4 + 2] / [3 + 2] addition pathway in kcal/mol, versus electrophilicity indices of the various acetylene derivatives
Regioselectivity
Along the [4 + 2]/[3 + 2] addition pathway, there are two possible regioisomers each arising from the reaction of 4Int and C. There are 1- and 6-regioisomers where 1 and 6 denote the positions C1-C2 and C6-C7 respectively on the 4Int-Ester where the incoming carbon of the diazopropane (C) may attack to form the transition states and the cycloadduct as shown in Scheme 3 and Fig. 8. For the [4 + 2]/[3 + 2] addition of dimethyl but-2-ynedioate with cyclopentadiene and 2-diazopropane, we found the 1-regioisomer to be unfavorable due to relatively high activation barriers (10.3 kcal/mol and 10.4 kcal/mol for the 4TSBExo-Ester and 4TSBEndo-Ester, respectively. However, along the 6-regioisomer pathway, the two possible transition states were found to have activation barriers of − 0.2 kcal/mol and 1.6 kcal/mol for the 4TSAExo-Ester and 4TSAEndo-Ester, respectively (see Table 1). Thus, clearly the formation of the 6-regioisomers would be favored and this accounts for the complete regioselectivity in this reaction.

Labels of atoms in the 4Int-Ester, 3Int-Ester, and C
To explain the origins of the regioselectivity in this reaction, we invoked perturbation molecular orbital (PMO) theory as employed in a similar study elsewhere [36]. The orbital energy obtained for the HOMOdipole (C) – LUMOdipolarophile (4Int-Ester) interaction is 5.79 eV and that of HOMOdipolarophile – LUMOdipole is 8.56 eV. Hence, the dominating orbital interactions will take place between the HOMO of the dipole and the LUMO of the dipolarophile since our calculations show that it is the interaction with closest energy, implying a normal electronic demand cycloaddition reaction.
Also, upon natural bond order (NBO) analysis, the molecular orbital coefficient of C1 and N2 in structure C are + 0.009 and − 0.052 respectively. Similarly, orbital coefficients of the dipolarophile have higher magnitude at the ester substituted olefinic carbons (C6 = − 0.22 and C7 = − 0.214) while that of the unsubstituted olefinic bond have lower magnitude (C1 = − 0.067 and C2 = − 0.082). By the rules of PMO theory, the cycloaddition will happen in a manner to unite the atoms with the highest molecular orbital coefficients since this would lead to the greatest stabilization [37]. Therefore, this accounts for the selectivity in favor of the ester-substituted olefinic bond. In the case of cyclopropane-substituted acetylene, we found the exo selective addition across the unsubstituted olefin to be the most favored path, and this can be attributed to steric effects.
Stereo- and chemo-selectivities
Again, at the stereoselective step along the [4 + 2] / [3 + 2] pathway, one would expect that the formation of the endo diastereomer will be preferred since it is a normal electron demand 1,3 – dipolar cycloaddition. Surprisingly, the exo diastereomer is observed to be favored. Using perturbation molecular orbital theory, the preference for the exo isomer can be rationalized.
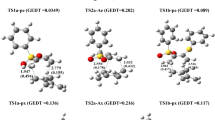
Assessment of the HOMO – LUMO gaps of TS2AExo-Ester and TS2AEndo-Ester show that the endo fashion (TS2AEndo-Ester) has a HOMO-LUMO gap of 7.12 eV (see Fig. 9a, b). In the case of the exo attack (TS2AExo-Ester), the HOMO-LUMO gap is lowered marginally to 6.98 eV (difference of 3.22 kcal/mol), a likely reason for the differences in free energy of activation for the formation of the two diastereomers.

Graphical depiction of the HOMO-LUMO plots of 4TS2AExo-Ester (a), 4TS2AEndo-Ester (b), 3TS2AEndo-Ester (c), and 3TS2AExo-Ester (d) with their orbital energies
Also, along the [3 + 2] / [4 + 2] addition pathway, we found the endo isomers to be favored in most cases. Upon PMO evaluation of all the substrates (see Fig. 9c, d), we found the 3TS2AEndo-Ester to have HOMO-LUMO gap of 6.90 eV while 3TS2AExo-Ester recorded 6.97 eV, a difference of 1.6 kcal/mol in favor of the endo; although the exo isomers were observed to have high thermodynamic stabilities over the endo.
None of the substrates were found to be favored towards a chemoselective addition across the N=N linkage of the pyrrole intermediate due to high activation barriers.
Conclusions
The mechanism of the reactions of functionalized acetylenes with cyclopentadiene and dimethyl diazopropane have been elucidated using density functional theory calculations and perturbation molecular orbital (PMO) theory arguments. We have demonstrated that in the reaction of unsubstituted acetylene with cyclopentadiene and dimethyl diazopropane, the sequence of the addition has no significant effects on product outcomes. The same product is expected to be obtained provided the reaction conditions remains the same for both [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] tandem addition sequences. It has also been established that the regio-, stereo-, and chemo-selectivities of the reaction are strictly dictated by the type of substituent on the parent acetylene. For substituted acetylenes, we conclude that the sequence of the tandem addition generally affects the type of isomeric product obtained. The [4 + 2]/[3 + 2] tandem addition sequence has been established to generally favor formation of the exo stereo-selective isomer across the substituted olefinic bond in the dipolarophile intermediate than the endo, whereas the [3 + 2]/[4 + 2] tandem addition sequence generally favors the corresponding endo product formation. It has also been found that the [4 + 2] Diels–Alder addition step irrespective of the addition sequence is the rate-determining step. The reaction of electron-donating substituted acetylenes along the [4 + 2]/[3 + 2] pathway has been found to generally increase the activation barriers compared to the parent acetylene. For electron-withdrawing substituted acetylenes, along the [4 + 2]/[3 + 2] pathway, we observed a decrease in activation energies in all the substituents except CH3, NH2, and Cl. Ring-substituted acetylene showed analogous energetic trends as that of the electron-releasing groups.
In addition, global reactivity indices (global electrophilicities and maximum electronic charge transfer) calculated for all the substituted-ethynes considered in this work show a good correlation with the activation energies.
References
Trost B M, Bartmann, W (1984) Selectivity-a Goal for Synthetic Efficiency: Proceedings of the 14. Workshop Conference Hoechst, Schloss Reisensburg 18–22 September, 1983. Verlag Chemie
Denmark SE, Thorarensen A (1996) Tandem [4+ 2]/[3+ 2] cycloadditions of nitroalkenes. Chem. Rev. 96(1):137–166
Bertz SH (1982) Convergence, molecular complexity, and synthetic analysis. J. Am. Chem. Soc. 104(21):5801–5803
Posner GH (1986) Multicomponent one-pot annulations forming 3 to 6 bonds. Chem. Rev. 86(5):831–844
Danishefsky S, Schuda PF, Kitahara T, Etheredge SJ (1977) The total synthesis of dl-vernolepin and dl-vernomenin. J. Am. Chem. Soc. 99(18):6066–6075
Neumann M F, Buchecker C D, Martina D, (1978) Tandem [4 + 2]/[3 + 2] and [3 + 2]/[4 + 2] addition reaction of cyclopentadiene with dimethyl acetylenedicarboxylate and a diazoalkane. J Chem Res (S). 78
Denmark SE, Baiazitov RY (2006) Tandem double-intramolecular [4+ 2]/[3+ 2] cycloadditions of nitroalkenes. Studies toward a total synthesis of daphnilactone B: piperidine ring construction. J Org Chem 71(2):593–605
Denmark SE, Baiazitov RY, Nguyen ST (2009) Tandem double intramolecular [4+ 2]/[3+ 2] cycloadditions of nitroalkenes: construction of the pentacyclic core structure of daphnilactone B. Tetrahedron 65(33):6535–6548
Denmark SE, Donald SM (1998) Tandem inter [4+ 2]/intra [3+ 2] cycloadditions. 17. The Spiro mode. Efficient and highly selective synthesis of azapropellanes. J Org Chem 63(5):1604–1618
de los Santos JM, Lopez Y, Aparicio D, Palacios F (2008) A convenient synthesis of substituted pyrazolidines and azaproline derivatives through highly regio-and diastereoselective reduction of 2-pyrazolines. J Org Chem 73(2):550–557
Sears JE, Barker TJ, Boger DL (2015) Total synthesis of (−)-vindoline and (+)-4-epi-vindoline based on a 1, 3, 4-oxadiazole tandem intramolecular [4+ 2]/[3+ 2] cycloaddition cascade initiated by an allene dienophile. Org. Lett. 17(21):5460–5463
Sears JE, Boger DL (2016) Tandem intramolecular Diels–Alder/1, 3-dipolar cycloaddition cascade of 1, 3, 4-oxadiazoles: initial scope and applications. Accs. Chem Res 49(2):241–251
Lu LQ, Li F, An J, Zhang JJ, An XL, Hua QL, Xiao WJ (2009) Construction of fused heterocyclic architectures by formal [4+ 1]/[3+ 2] cycloaddition cascade of sulfur ylides and nitroolefins. Ange Chem 121(50):9706–9709
Fox ME, Li C, Marino JP, Overman LE (1999) Enantiodivergent total syntheses of (+)-and (−)-scopadulcic acid a. J. Am. Chem. Soc. 121(23):5467–5480
Nicolaou KC, Montagnon T, Snyder SA (2003) Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem Comm (5) 551–564.
Paton RS, Steinhardt SE, Vanderwal CD, Houk KN (2011) Unraveling the mechanism of cascade reactions of Zincke aldehydes. J. Am. Chem. Soc. 133(11):3895–3905
Chawla R, Sahoo UL, Arora A, Sharma PC, Radhakrishnan V (2010) Microwave-assisted synthesis of some novel 2-pyrazoline derivatives as possible antimicrobial agents. Acta Pol. Pharm. Drug Res. 67(1):55–61
Chimenti F, Bizzarri B, Manna F, Bolasco A, Secci D, Chimenti P, Brenciaglia MI (2005) Synthesis and in vitro selective anti-Helicobacter pylori activity of pyrazoline derivatives. Bioorg. Med. Chem. Lett. 15(3):603–607
Sivakumar PM, Ganesan S, Veluchamy P, Doble M (2010) Novel chalcones and 1, 3, 5-triphenyl-2-pyrazoline derivatives as antibacterial agents. Chem Bio Drug Des 76(5):407–411
Wavefunction, Inc. (2013) Spartan ‘14. Wavefunction, Inc., Irvine
Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41(2):157–167
Pieniazek SN, Clemente FR, Houk KN (2008) Sources of error in DFT computations of C=C bond formation thermochemistries: π→σ transformations and error cancellation by DFT methods. Ang Chem Int Ed 47(40):7746–7749
Pieniazek SN, Houk KN (2006) The origin of the halogen effect on reactivity and reversibility of Diels–Alder cycloadditions involving furan. Ang Chem Int Ed 45(9):1442–1445
Paton RS, Mackey JL, Kim WH, Lee JH, Danishefsky SJ, Houk KN (2010) Origins of stereoselectivity in the trans Diels–Alder paradigm. J. Am. Chem. Soc. 132(27):9335–9340
Paton RS, Kim S, Ross AG, Danishefsky SJ, Houk KN (2011) Experimental Diels–Alder reactivities of cycloalkenones and cyclic dienes explained through transition-state distortion energies. Ang Chem Int Ed 50(44):10366–10368
Wheeler SE, Moran A, Pieniazek SN, Houk KN (2009) Accurate reaction enthalpies and sources of error in DFT thermochemistry for aldol, Mannich, and α-aminoxylation reactions. J. Phys. Chem. A 113(38):10376–10384
Chmely SC, Kim S, Ciesielski PN, Jiménez-Osés G, Paton RS, Beckham GT (2013) Mechanistic study of a Ru-Xantphos catalyst for tandem alcohol dehydrogenation and reductive aryl-ether cleavage. ACS Cat 3(5):963–974
Clark M, Cramer RD, Van Opdenbosch N (1989) Validation of the general purpose Tripos 5.2 force field. J Comp Chem 10(8):982–1012
Opoku E, Tia R, Adei E (2016) [3 + 2] versus [2 + 2] Addition: A Density Functional Theory Study on the Mechanistic Aspects of Transition Metal-Assisted Formation of 1,2-Dinitrosoalkanes. Journal of Chemistry 2016:10
Fosu E, Tia R, Adei E (2016) Mechanistic studies on Diels–Alder [4+ 2] cycloaddition reactions of α, β-substituted cyclobutenones: role of substituents in regio-and stereoselectivity. Tetrahedron 72(50):8261–8273
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem. Rev. 105(8):2999–3094
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels–Alder reactions. J. Phys. Chem. A 106(29):6871–6875
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J. Am. Chem. Soc. 121(9):1922–1924
Koopmans T (1934) Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1(1–6):104–113
Nantogma S, Tia R, Adei E (2018) A DFT mechanistic study of the generation of azomethine ylides from the ring-opening reactions of stabilized aziridines and follow-up 1, 3-dipolar cycloaddition reactions with acetaldehyde. Comp Theo Chem 1144:38–49
Houk KN (1975) Frontier molecular orbital theory of cycloaddition reactions. Acc. Chem. Res. 8(11):361–369
Acknowledgements
The authors are very grateful to the National Council for Tertiary Education, Republic of Ghana, for a research grant under the Teaching and Learning Innovation Fund (TALIF/KNUST/3/0008/2005), and to South Africa’s Centre for High Performance Computing for access to additional computing resource.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no conflicts of interest regarding this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Opoku, E., Tia, R. & Adei, E. Computational studies on [4 + 2] / [3 + 2] tandem sequential cycloaddition reactions of functionalized acetylenes with cyclopentadiene and diazoalkane for the formation of norbornene pyrazolines. J Mol Model 25, 168 (2019). https://doi.org/10.1007/s00894-019-4056-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4056-x