
Abstract
The effects of the molar ratio, temperature, and solvent on the formation of the cocrystal explosive DNP/CL-20 were investigated using molecular dynamics (MD) simulation. The cocrystal structure was predicted through Monte Carlo (MC) simulation and using first-principles methods. The results showed that the DNP/CL-20 cocrystal might be more stable in the molar ratio 1:1 near to 318 K, and the most probable cocrystal crystallizes in the triclinic crystal system with the space group P\( \overline{1} \). Cocrystallization was more likely to occur in methanol and ethanol at 308 K as a result of solvent effects. The optimized structure and the reduced density gradient (RDG) of the DNP/CL-20 complex confirmed that the main driving forces for cocrystallization were a series of hydrogen bonds and van der Waals forces. Analyses of the trigger bonds, the charges on the nitro groups, the electrostatic surface potential (ESP), and the free space per molecule in the cocrystal lattice were carried out to further explore their influences on the sensitivity of CL-20. The results indicated that the DNP/CL-20 complex tended to be more stable and insensitive than pure CL-20. Moreover, an investigation of the detonation performance of the DNP/CL-20 cocrystal indicated that it possesses high power.

DNP/CL-20 cocrystal models with different molar ratios were investigated at different temperatures using molecular dynamics (MD) simulation methods. Binding energies and mechanical properties were probed to determine the stability and performance of each cocrystal model. Solvated DNP/CL-20 models were established by adding solvent molecules to the cocrystal surface. The binding energies of the models in various solvents were calculated in order to identify the most suitable solvent and temperature for preparing the cocrystal explosive DNP/CL-20
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
When two or more neutral species are combined via intermolecular interactions in a certain ratio, new cocrystals are created with distinct physicochemical properties [1, 2]. For some high explosives, such as 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20), the sensitivity can be modified without sacrificing much of the detonation energy through cocrystallization [3,4,5]. Hence, cocrystal explosives, especially energetic–energetic cocrystals, have attracted a great deal of attention in the field of energetic materials.
Although cocrystal explosives have been proposed for many years, only a few CL-20-based cocrystal explosives have been synthesized experimentally due to the exacting experimental conditions required. For instance, the cocrystal explosive CL-20/TNT [3, 6] was prepared in the molar ratio 1:1 by the slow evaporation of ethanol and ethyl acetate. The cocrystal CL-20/1,3,5,7-tetranitro-1,3,5,7-tetrazacyclooctane (HMX) [7] in the molar ratio 2:1 was prepared in 2-propanol. CL-20/benzotrifuroxan (BTF) [8] was prepared in several saturated organic solvents in the molar ratio 1:1, and high-quality crystals were obtained from ethanol. CL-20/1,3-dinitrobenzene (DNB) in the molar ratio 1:1 [9] was obtained in ethanol at 30 °C. Bimolecular crystals of CL-20 with 2,4-dinitro-2,4-diazapentane in the molar ratio 2:1 and CL-20 with 2,4-dinitro-2,4-diazaheptane in the molar ratio 1:1 were obtained in ethyl ethanoate at room temperature [10]. CL-20/1,3,5-triamino-2,4,6-trinitrobenzene (TATB) [11] in the molar radio 3:1 was prepared by a rapid solvent/no-solvent method. CL-20/2,5-dinitrotoluene (DNT) [12] in the molar ratio 1:2 was prepared in ethyl acetate at room temperature. CL-20/cyclotrimethylenetrinitramine (RDX) in the molar ratio 1:1 was prepared in acetonitrile [13]. Some CL-20 cocrystals containing nonenergetic materials have also been prepared [5]. Despite the rather brief reports of those laboratory studies that are available, it is evident that the molar ratio, the solvent, and the temperature play crucial roles in cocrystal growth. Indeed, theoretical studies performed by our group indicate that the binding energies, sensitivities, and detonation performance of cocrystal explosives are influenced by the molar ratio of the cocrystal [14,15,16]. Differences in solubility parameters have been studied in order to screen for cocrystal formers using quantum chemical calculations [17]. The effects of the solvent acetonitrile on diacetone diperoxide (DADP)/1,3,5-tribromo-2,4,6-trinitrobenzene (TBTNB) cocrystallization were investigated, and the results highlighted that it is important to consider the influences of the temperature and the solvent when designing new cocrystals [18].
While many cocrystal explosives have been studied using experimental and theoretical methods, only a few studies of energetic azole-based cocrystals have been reported besides our theoretical investigation of the HMX/1-methyl-4,5-dinitroimidazole (MDNI) cocrystal [19]. 3,4-Dinitropyrazole (DNP) is a novel insensitive pyrazole explosive that is a potential substitute for the melt-cast explosive 2,4,6-trinitrotoluene (TNT) [20]. However, the cocrystal DNP/CL-20 may have better properties than pure DNP due to the low sensitivity of DNP, although neither experimental nor theoretical studies of the DNP/CL-20 cocrystal have been reported in the literature thus far. Therefore, in the work summarized in the present paper, the influences of the molar ratio and the solvent on this pyrazole/nitroamine cocrystal at different temperatures were explored. These influences were gauged by calculating binding energies and mechanical properties. The intermolecular interactions between DNP and CL-20 were investigated using density functional theory (DFT), employing the B3LYP, M062X, and B97D methods. A series of trigger bond analyses were implemented to study the impacts of intermolecular interactions on the sensitivity of CL-20. The structure of the DNP/CL-20 cocrystal was predicted in order to evaluate its detonation performance. Temperature-dependent solvent effects were studied using SMD models and molecular dynamics (MD) simulation. These theoretical results for the cocrystal explosive DNP/CL-20 should prove useful in attempts to prepare novel pyrazole/nitroamine cocrystal explosives.
Computational methods
Molecular dynamics calculations
In the present work, ε-CL-20 was chosen as the initial configuration of the DNP/CL-20 cocrystal due to its excellent performance. The unit cells of DNP and CL-20 were obtained from [21, 22]. COMPASS [23] was found to be a reliable force field for predicting the structural and thermophysical properties of cocrystal explosives in our previous studies [14,15,16, 20, 24, 25]. The crystal structures of DNP and CL-20 are illustrated in Fig. 1. Supercells of DNP and CL-20 were constructed as a means to build initial DNP/CL-20 cocrystal models with molar ratios of 2:1, 3:2, 1:1, 2:3, and 1:2. The CL-20 layer was positioned below the DNP layer, and the vacuum slab thicknesses above the CL-20 layer and the DNP layer were set to be 10 Å and 60 Å, respectively. The corresponding parameters (molar ratio, supercells, total number of molecules, the number of DNP (N DNP) units, and the number of CL-20 (N CL-20) units) are given in Table 1.

Crystal structures of DNP and CL-20
After building and optimizing the geometries of the cocrystal models, NVT ensembles and specific temperatures (298, 308, 318, 328, and 338 K) were employed to obtain the ideal models using MD simulation. Andersen’s [26] temperature control method was selected. The time step was set to 2.0 fs, and the total dynamic time was 200.0 ps (200,000 fs). The binding energy (E bind) and the mechanical properties of the equilibrium state of DNP/CL-20 were then calculated to evaluate their compatibility. To predict the crystal structure of DNP/CL-20, the seven most likely space groups (C2/c, P\( \overline{1} \), P21/c, P21, P212121, Pbca, and Pna21) were compared using the COMPASS force field and Monte Carlo (MC) simulation.
Furthermore, the effects of the solvent on the DNP/CL-20 cocrystal were simulated by adding methanol to the cocrystal surface. The unit cells of the DNP/CL-20 cocrystal and methanol were obtained from the crystal structure predicted by MC simulation and from [27], respectively. The supercells of the DNP/CL-20 cocrystal and methanol were utilized to build an initial model of methanol-solvated DNP/CL-20. After optimization, the methanol supercell layer (including 48 molecules) was positioned above the cocrystal layer (2 × 2 × 1 supercell), and the vacuum slab thickness above the cocrystal layer was set at 5 Å. NVT-MD using the Andersen method was employed to acquire the ideal model. The time step was set to 1.0 fs and the total dynamic time was 100.0 ps (100,000 fs). Binding energies were calculated to evaluate the stability of the model at 298, 308, 318, 328, and 338 K, respectively. All of these calculations were carried out with MS 7.0 [28].
Quantum chemical calculations
Four structures of DNP/CL-20 were designed in order to study the intermolecular interactions of the cocrystal. Those structures were optimized at the B3LYP level [29] with the 6–311++G(d,p) basis set. Local minima were checked to ensure that there were no imaginary frequencies. Interaction energies were computed using the M06-2X [30] and B97D [31] methods with the Boys–Bernardi counterpoise procedure [32]. The sensitivity of CL-20 was explored by analyzing trigger bonds, nitro group charges, electrostatic surface potential (ESP), and the free space per molecule in the cocrystal lattice. The reduced density gradient (RDG) was also used to study the nature of the intermolecular interactions. All studies were executed in the Gaussian 09 [33] and Multiwfn software packages [34].
Results and discussion
Binding energy analysis
The binding energies of the various DNP/CL-20 cocrystal models were calculated and the results are listed in Table 2. They were found to lie within the range 604.00–1126.45 kJ/mol, and the binding energy of the 1:1 model at 318 K was the largest among all of the models considered. Plots of molar ratio and temperature vs. binding energy \( {E}_{\mathrm{bind}}^{\ast } \) are depicted in Fig. 2, and show specific trends. In the plot of molar ratio vs. binding energy, the binding energy clearly increases as the molar ratio shifts from 2:1 to 1:1, except at 318 K. When the amount of DNP surpasses that of CL-20, the binding energy starts to get smaller, highlighting the pivotal roles of the molar ratio and the temperature in cocrystal formation. From the plot of temperature vs. binding energy (the right curve in Fig. 2), the largest binding energies for the 2:1, 3:2, 1:1, 2:3, and 1:2 models can be seen to occur at 318, 308, 318, 328, and 328 K, respectively, corresponding to binding energies of 993.02, 1031.30, 1126.45, 947.40, and 666.18 kJ/mol. The binding energy of the 1:1 model is the largest at various temperatures, indicating that the strongest intermolecular interactions occur in the 1:1 model at each temperature. The binding energy of the 1:2 model is the smallest at each temperature, ranging from 604.00 to 666.18 kJ/mol. The binding energy of the cocrystal is regarded as an indication of its stability: the larger the binding energy, the more stable the cocrystal. Therefore, the optimal molar ratio for the DNP/CL-20 cocrystal is 1:1, and the optimal temperature for the growth of such a cocrystal is near to 318 K in vacuum.

Plots of the molar ratio and temperature versus the binding energy \( {E}_{\mathrm{bind}}^{\ast } \) for the DNP/CL-20 cocrystal
Mechanical properties
Mechanical properties play an important role in the preparation, machining, and storage of energetic materials. Elastic stiffness constants (C ij ) and elastic moduli were used to evaluate the plastic deformation and tensile strength of the DNP/CL-20 cocrystal. K/G and the Cauchy pressure (C 12 – C 44) are indicators of the ductility of the material, with more positive values implying better ductility.
The mechanical parameter values of the cocrystal models with different molar ratios are listed in Table 3. The DNP/CL-20 cocrystal shows anisotropic behavior given that the coefficients C ij differ in value (see Table S1 in the “Electronic supplementary material,” ESM). Based on the elastic modulus (K, G, and E) values for the different models, the rigidity decreases (while the elasticity increases) in the following order: 2:3 > 1:1 > 1:2 > 3:2 > 2:1. DNP/CL-20 cocrystallization weakens the influence of environmental stimuli on the CL-20, which in turn decreases the probability of hot spot formation, implying that the DNP/CL-20 crystal is less sensitive than pure CL-20. The value of C 12 − C 44 is positive for all the models, suggesting that the ductility of the DNP/CL-20 cocrystal is satisfactory. A material is usually considered to show good plasticity if the value of Poisson’s ratio for the material lies between 0.2 to 0.4. However, Poisson’s ratio for the 2:3 model is 0.029, which is the lowest among all the models tested. The value of K/G for the 2:3 model is also the lowest among all the models.
In summary, though not all the mechanical property values of the 1:1 cocrystal model are the best among all of the models, overall, the 1:1 model affords the best mechanical properties. Therefore, given the results of binding energy analysis, we focused on the 1:1 cocrystal model in subsequent investigations.
Structure analysis
Four possible structures of the DNP/CL-20 complex and some important parameter values for them are shown in Fig. 3, all of which were optimized at the B3LYP/6–311++G(d,p) level. All the structures possess C1 symmetry. According to Fig. 3, the main intermolecular interactions are between nitrogens of nitro groups and hydrogens of C–H groups. The lengths of the H···O/N bonds are 2.08–2.62 Å and the angles of the C–H···O/N bonds are 121.9–167.3°. All of the H···O/N bond lengths are within the generally accepted range of hydrogen-bond lengths [36], and the H···O and H···N lengths are shorter than the sum of the van der Waals radii of the hydrogen atom (1.20 Å) and either the oxygen atom (1.52 Å) or the nitrogen atom (1.55 Å) [35]. Moreover, the shorter the H···A distance, the closer the D–H···A angle is to 180°, and the stronger the hydrogen bond [37]. Therefore, intermolecular hydrogen-bonding interactions may exist in the DNP/CL-20 complex, and the interactions in structures II and III are the strongest among all the structures.

Optimized structures of the DNP/CL-20 complex calculated at the B3LYP/6–311++G(d,p) level. Gray dotted lines indicate intermolecular interactions. Bond lengths and bond angles are also shown. C, H, O, and N atoms are shown in gray, white, red, and blue, respectively
Energy and stability
The interaction energy E(int.), defined as the difference in energy between the complex and the isolated monomers, was also investigated. Table 4 lists uncorrected and corrected interaction energies for the four structures of the DNP/CL-20 complex calculated by applying the 6–311++G(d,p) and 6–311++G(2df,2p) basis sets and the M06-2X and B97D methods to their fully optimized geometries. It is known that the B3LYP method describes the bond dissociation energy (BDE) better than the MP2 method does [38]. Therefore, the BDEs of the trigger bonds were obtained at the B3LYP/6–311++G(d,p) level. The Laplacian bond order [39] (LBO) and the electron density at each BCP were calculated to probe the strength of each trigger bond. The charge on a nitro group Q(NO2) was employed to evaluate the sensitivities of the complexes: the more negative the charge on a nitro group, the lower the sensitivity of the explosive [40]. The calculated Q(NO2) values (atomic dipole moment corrected (ADCH) Hirshfeld atomic charges [41]) are listed in Table 6.
As listed in Table 4, the uncorrected interaction energies are—as expected—smaller than the corrected energies, due to the basis set superposition error. It can be seen that the E(int.) values of the four structures lie within the range −160 to −16 kJ/mol, which suggests that they are intermolecular hydrogen-bonding interactions [42]. The interaction energies of the four structures can be ordered as follows: II ≈ III < IV < I, meaning that structures II and III are more stable than the other two.
As shown in Table 5, upon comparing the four complexes with the isolated CL-20 molecule, the lengths of N–N bonds in the complexes were found to be 0.005–0.015 Å shorter, the BDEs of N–N bonds in the complexes were found to be 6.1–20.9 kJ/mol higher, the LBOs of N–N bonds in the complexes were found to be 0.02–0.07 greater, and the electron density at the BCPs of N–N bonds in the complexes were found to be 0.004–0.0133 a.u. higher. Also, from Fig. 4, there are strong linear relationships between the change in the BDE, the change in the bond length, the change in the bond order, and the change in ρ BCP for trigger bonds.

Plots of a ΔBDE vs. change in bond length and Δρ BCP, b change in bond order vs. ΔBDE and Δρ BCP, and c change in bond length vs. change in bond order and Δρ BCP for the trigger bonds of the four DNP/CL-20 complexes
In addition, these trigger bonds are all in direct contact with an atom involved in intermolecular interactions, indicating that intermolecular interactions change trigger bond strength and influence the sensitivity of the complex. In the four DNP/CL-20 structures, CL-20 is less sensitive than isolated CL-20. In particular, for CL-20 in structure III, the trigger bond is 0.015 Å longer, the BDE is 20.9 kJ/mol larger, the bond order is 0.07 greater, and the electron density is 0.0133 a.u. higher than in isolated CL-20.
In Table 6, it is clear that the atomic charges on the nitro groups in the complexes are more negative than those in the CL-20 monomer. For example, the Q(NO2) of the N(32)O(35)O(36) nitro group in the monomer is −0.04098e, but it is −0.0880e and −0.1342e for structures I and III, respectively. Results show that the charges on the CL-20 nitro groups become more negative with the formation of intermolecular hydrogen-bonding interactions, which decreases the sensitivity of CL-20 to some extent.
RDG analysis
Yang et al. [43] developed a method of detecting noncovalent interactions in real space, based on the reduced density gradient (RDG) and the electron density multiplied by the sign of the second Hessian eigenvalue (sign(λ 2)ρ). Sign(λ 2)ρ can be used to distinguish bonded (λ 2 < 0) from nonbonded (λ 2 > 0) interactions. The RDG is defined as \( \mathrm{RDG}=\frac{1}{2{\left(3{\pi}^2\right)}^{1/3}}\frac{1}{2{\left(3{\pi}^2\right)}^{1/3}} \).
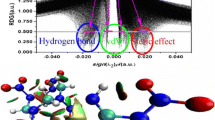
To visually investigate weak intermolecular interactions, scatter diagrams and isosurface graphs of the RDG for various regions of the complexes were plotted, as shown in Fig. 5. In the scatter diagrams, the x axis is the function sign(λ 2)ρ and the y axis is the RDG, and the function sign(λ 2)ρ ranges from −0.05 a.u. to 0.05 a.u. Also, the density of points corresponds to the electron density (i.e., a high point density indicates a high electron density).

Scatter diagrams and isosurface graphs of the RDG for the four DNP/CL-20 structures I–IV. The structures in the isosurface graphs are colored according to a blue-green-red scale that correlates with the value of sign(λ 2)ρ in the range −0.04 to 0.02 a.u.
Looking at the scatter diagrams, we can see some spikes, which can be classified into three types indicated by dark green, pale green, and red circles in the diagrams; these spikes represent hydrogen-bonding, van der Waals (vdW)-force, and steric regions, respectively. These three sets of spikes occur around (λ 2)ρ = −0.02, −0.01, and < 0 a.u., respectively. In the RDG isosurface graphs, there are elliptical dark green slabs located at hydrogen, oxygen, or nitrogen atoms between the DNP and CL-20, indicating that there are strong vdW and weak hydrogen-bonding interactions at these locations. Moreover, among the four structures, structure III exhibits the strongest and the most spikes in the attractive region. Spikes in the sign(λ 2)ρ < 0 region suggest the presence of steric effects, which correspond to the red regions in CL-20 and DNP. Note that blue isosurfaces are observed in the region between the nitro groups in DNP and CL-20 in complex II, whereas blue isosurfaces are found in the region between the nitro groups of CL-20 and the ring nitrogen of DNP in complex III. This implies that there are O···O and O···N interactions but not H···O/N interactions in complexes II and III. Indeed, according to atoms-in-molecules (AIM) [44] analysis (Table S2 of the ESM), the electron densities of O(44)···N(29), O(44)···O(36), and O(10)···O(46) are 0.0039, 0.0057, and 0.0043 a.u., respectively.
Electrostatic potential analysis
The electrostatic surface potential (ESP) was considered when analyzing the sensitivities of the four DNP/CL-20 complexes. The ESPs of CL-20 and complexes I–IV were calculated based on the corresponding structures optimized at the B3LYP/6–311++G(d,p) level. Results and figures relating to the ESP are shown in Tables S3–S7 and Figs. S1–S5 of the ESM. The values of \( {\sigma}_{+}^2 \), \( {\sigma}_{-}^2 \) and \( \nu ={\sigma}_{+}^2{\sigma}_{-}^2/{\left({\sigma}_{+}^2+{\sigma}_{-}^2\right)}^2 \) obtained from Tables S3–S7 of the ESM are listed in Table 7. Politzer reported that the smaller the value of \( {\sigma}_{+}^2 \) and the larger the value of ν, the lower the impact sensitivity of the explosive [45]. From Table 7, although complex I has a larger value of \( {\sigma}_{+}^2 \) than CL-20 does, complexes II, III, and IV all have smaller values than CL-20 does. Meanwhile, the ν values of complexes II, III, and IV are larger than that of CL-20. Therefore, the impact sensitivity of the DNP/CL-20 cocrystal may be lower than that of CL-20. However, it is important to note that the sensitivity of an explosive is controlled by a variety of factors, not just ν [46].
Crystal structure prediction
The lattice energy is considered an important stability criterion for cocrystals [47]. Crystal density and cell parameter values predicted by MC simulations of seven possible space groups (labeled 1–7, respectively) for the DNP/CL-20 cocrystal are listed in Table 8. The smaller the lattice energy, the greater the stability, so the triclinic crystal system with space group P\( \overline{1} \) appears to be the most likely structural system for the DNP/CL-20 cocrystal (see Fig. 6). The cell parameter values for this space group are a = 12.24 Å, b = 14.14 Å, c = 7.51 Å, α = 100.80°, β = 107.39°, γ = 116.96°, Z = 2, and ρ = 1.937 g/cm3. A slight decrease in density compared to that of CL-20 was observed for all possible crystal structures except for the orthorhombic crystal system with space group Pbca. This is similar to what was found in a study of TNT cocrystals, with some cocrystals showing higher density than TNT upon cocrystallization [48].

The most likely crystal structure (left) and crystal morphology (right) of the DNP/CL-20 cocrystal
Free space per molecule in the cocrystal lattice
The conversion of mechanical energy into hot spots and the scission of trigger bonds can occur relatively easily if there is considerable free space in the crystal lattice [49, 50]. Consequently, the free space per molecule in the cocrystal lattice can be a useful parameter to study in relation to the sensitivity of explosives. In our work, each DNP/CL-20 cocrystal framework 1–7 predicted by MC stimulations was regarded as a cocrystal “molecule.” Therefore, the free space in DNP/DNP and CL-20/CL-20 dimers was examined in order to evaluate the sensitivity of the cocrystal. The free space values of the cocrystal molecules corresponding to the seven space groups (structures 1–7), a single DNP molecule, a single CL-20 molecule, and the DNP/DNP and CL-20/CL-20 dimers were calculated using Eqs. 1 and 2 below, and the results are listed in Table 9.
where ΔV, V eff, and V int represent the free space per molecule in the unit cell, the effective volume per molecule, and the intrinsic molecular volume, respectively. V int is the molecular volume encompassed by the 0.003 a.u. electron density contour.
where M and ρ are the molecular mass and the crystal density, respectively.
From Table 9, it is clear that the ΔV values for the cocrystal molecules are higher than that of the DNP dimer but lower than that of the CL-20 dimer, indicating that there is less free space in the cocrystal than in the CL-20 dimer, but more free space in the cocrystal than in the DNP dimer. The ΔV values of cocrystals 1, 2, and 7 are larger than those of cocrystals 3, 4, 5, and 6, suggested that there is more free space in the former set of cocrystals than in the latter. Studiees [49,50,51,52] have demonstrated that the larger the free space per molecule (ΔV), the higher the sensitivity of the cocrystal. Therefore, we can predict that DNP/CL-20 is less sensitive than CL-20 but more sensitive than DNP. For these possible DNP/CL-20 cocrystal frameworks, the free space per “molecule” in the cocrystal lattice is large if the density is low. As mentioned above, it is difficult to accurately predict the sensitivity of an explosive based on one parameter. Hence, while trends in explosive sensitivity can be predicted, it is not possible to closely correlate the sensitivity with any particular factor.
Analysis of the effect of the solvent
In our laboratory, cocrystal explosives based on CL-20 were prepared by slow solvent evaporation. In order to study the influence of the solvent on the stability of the DNP/CL-20 cocrystal, the SMD model [53] was utilized to study the interaction energies of the four DNP/CL-20 complexes I–IV with five solvents at the M06-2X/6–311 + G(d,p) level (see Table 10). The solvents were selected after careful consideration. On the one hand, these solvents are often used to prepare cocrystals in labs; for example, methanol has been used to prepare the NTO/TZTN cocrystal [54]. On the other hand, in order to explore the effect of the solvent using the SMD model, we chose five solvents—ethyl acetate, dichloroethane, acetone, ethanol, and methanol—that cover a wide range of dielectric constants: 6.1, 10.4, 20.7, 24.3, and 32.6 D, respectively.
According to Table 10, whichever complex and solvent combination is considered, the magnitude of the interaction energy decreases by at least 20% when solvent effects are taken into account. Among all five solvents, the interaction energies for the complexes in methanol are the most negative. This result contrasts with the results of a study of HMX/Fox-7 [55], where the interaction energies were found to decrease as the dielectric constant of the solvent increased from 5.0 to 50.0 D. As discussed above, complex II is the most stable structure of the DNP/CL-20 cocrystal, and the magnitude of the interaction energy was found to decrease least in the solvents methanol and ethanol. Therefore, methanol and ethanol appear to be the best solvents for preparing the DNP/CL-20 cocrystal.
However, the effects of the solvent comprise far more than simply the effects of the solvent’s dielectric constant. Therefore, the effects of methanol and ethanol on the 1:1 DNP/CL-20 cocrystal were further simulated by adding solvent molecules to the cocrystal surface. Because there was little difference in interaction energy between the SMD models of methanol and ethanol, the effects of these solvents were also studied by MD stimulation. The crystal structure of DNP/CL-20 with the space group P\( \overline{1} \) was utilized to build a cocrystal supercell. The original solvated DNP/CL-20model is displayed in Fig. 7. As illustrated in Table 11 and Fig. 8, the values of E bind/solvent were found to be highest at 308 K in methanol and ethanol. The magnitude of the binding energy in either methanol or ethanol increases as the temperature rises from 298 to 308 K, but it decreases rapidly as the temperature rises further from 308 to 338 K. These results suggest that the temperature has a significant influence on the solvent effect for the cocrystal. In methanol and ethanol, the most suitable temperature for the crystallization of the DNP/CL-20 cocrystal is near to 308 K due to complex solvent effects. It is worth noting that the crystal-forming process is complicated, so some important parameters such as solubility and the rate of evaporation must be taken into account in cocrystal experiments.

Original model of DNP/CL-20 in methanol; the molecules in dark green are DNP

Plot of temperature T vs. binding energy \( {E}_{\mathrm{bind}/\mathrm{solvent}} \) for the 1:1 DNP/CL-20 cocrystal
Cocrystal properties
The detonation velocity V D, detonation pressure P D, detonation heat Q, and the specific impulse I sp were calculated for cocrystals 1–7 with different space groups, and are listed in Table 12. The V D values of these seven DNP/CL-20 cocrystal structures are 8.62, 8.82, 8.96, 8.95, 8.95, 9.19, and 8.48 km/s, respectively, which are a little lower than those of CL-20 but much higher than those of DNP. The P D values of the DNP/CL-20 cocrystals are also lower than that of CL-20. Moreover, there is a general tendency for sensitivity to increase as the detonation heat Q increases (although the correlation between these parameters is not strong) [58]. The Q values of the seven possible DNP/CL-20 cocrystals lie between 5.46 MJ/kg and 5.57 MJ/kg, which makes them lower than the Q value of CL-20 (5.79 MJ/kg) but higher than that of DNP (4.85 MJ/kg). On this basis, we would expect the sensitivity of DNP/CL-20 to be lower than that of CL-20 but higher than that of DNP. Therefore, although the detonation properties of the DNP/CL-20 cocrystal are slightly less impressive than those of pure CL-20, the cocrystal is less sensitive. Further, the specific impulse values of the DNP/CL-20 cocrystals are very similar to that of CL-20, demonstrating that DNP/CL-20 has a desirable energy efficiency of combustion. Indeed, the detonation performance of cocrystal 2, which is the most likely cocrystal structure, is pretty good (V D = 9.09 km/s, P D = 37.2 Gpa, Q = 5.52 MJ/kg).
Conclusions
Although there is no doubt that the molar ratio and effect of the solvent play non-negligible roles in the growth processes of cocrystal explosives, there are many unknown laws waiting to be explored. In this work, we investigated the influences of the molar ratio and temperature on the binding energy and mechanical properties of the DNP/CL-20 cocrystal, thus establishing their importance. An analysis of solvent effects also proved that the temperature and solvent are crucial factors to consider when preparing a cocrystal explosive. The chief conclusions of this work are listed below:
-
1.
The DNP/CL-20 cocrystal explosive is most likely to form at a molar ratio of 1:1 and 318 K in vacuum, and the resulting cocrystal is predicted to show good mechanical properties. An analysis of the effect of the solvent employed on the cocrystallization suggested that methanol and ethanol could be potential candidates for the preparation of the DNP/CL-20 cocrystal using a slow solvent evaporation method, and that the appropriate preparation temperature in methanol and ethanol is then near to 308 K.
-
2.
The interaction energies of the possible DNP/CL-20 complexes I–IV are predicted to be −24.18, −48.12, −46.78, and −37.61 kJ/mol, respectively, indicating that structures II and III are the most stable. RDG analysis indicated that the intermolecular interactions between DNP and CL-20 are dominated by CH···O and NH···O hydrogen bonds as well as O···O and N···O van der Waals forces; these results were supported by those from AIM analysis.
-
3.
Compared with the CL-20 monomer, the trigger bond is shorter; the bond order, BCP electron density, and BDE are larger; and the charge on a nitro group is more negative in DNP/CL-20 complexes, meaning that the formation of intermolecular interactions in these complexes make them more stable and less sensitive than the CL-20 monomer. Analyses of the ESP, the free space per molecule in the crystal lattice, and the heat of detonation showed that the impact sensitivity of DNP/CL-20 tends to be lower than that of pure CL-20.
-
4.
The DNP/CL-20 cocrystal is most likely to crystallize in a triclinic crystal system with space group P\( \overline{1} \). The corresponding cell parameters are a = 12.24 Å, b = 14.14 Å, c = 7.51 Å, α = 100.80°, β = 107.39°, γ = 116.96°, Z = 2, and ρ = 1.937 g/cm3.
-
5.
The predicted velocity, pressure, and heat of detonation for the DNP/CL-20 cocrystal are 9.09 km/s, 37.2 GPa, and 5.52 MJ/kg, respectively.
References
Anslyn EV, Dougherty DA (2004) Modern physical organic chemistry. University Science Books, Sausalito
Lara-Ochoa F, Espinosa-Pérez G (2007) Cocrystals definitions. Supramol Chem 19:553–557
Bolton O, Matzger AJ (2011) Improved stability and smart-material functionality realized in an energetic cocrystal. Angew Chem Int Ed 50:8960–8963
Lin H, Zhu S-G, Li H-Z, Peng X-H (2013) Structure and detonation performance of a novel HMX/LLM-105 cocrystal explosive. J Phys Org Chem 26:898–907
Millar DIA, Maynard-Casely HE, Allan DR, Cumming AS, Lennie AR, Mackay AJ, Oswald IDH, Tang C-C, Pulham CR (2012) Crystal engineering of energetic materials: co-crystals of CL-20. Cryst Eng Comm 14:3742–3749
Yang Z-W, Li H-Z, Huang H, Zhou X-Q, Li J-S, Nie F-D (2013) Preparation and performance of a HNIW/TNT cocrystal explosive. Propell Explos Pyrot 38:495–501
Bolton O, Simke LR, Pagoria PF, Matzger AJ (2012) High power explosive with good sensitivity: a 2:1 cocrystal of CL-20:HMX. Cryst Growth Des 12:4311–4314
Yang Z-W, Li H-Z, Zhou X-Q, Zhang C-Y, Huang H, Li J-S, Nie F-D (2012) Characterization and properties of a novel energetic–energetic cocrystal explosive composed of HNIW and BTF. Cryst Growth Des 12:5155–5158
Wang Y-P, Yang Z-W, Li H-Z, Zhou X-Q, Zhang Q, Wang J-H, Liu Y-C (2014) A novel cocrystal explosive of HNIW with good comprehensive properties. Propell Explos Pyrot 39:590–596
Goncharov TK, Aliev ZG, Aldoshin SM, Dashko DV, Vasil AA, Shishov NI, Milekhin YM (2015) Preparation, structure, and main properties of bimolecular crystals CL-20-DNP and CL-20-DNG. Russ Chem B+ 64:366–374
Xu H-F, Duan X-H, Li H-Z, Pei C-H (2015) A novel high-energetic and good-sensitive cocrystal composed of CL-20 and TATB by a rapid solvent/non-solvent method. RSC Adv. 5:95764–95770
Liu K, Zhang G, Luan J-Y, Chen Z-Q, Su P-F, Shu Y-J (2016) Crystal structure, spectrum character and explosive property of a new cocrystal CL-20/DNT. J Mol Struct 1110:91–96
Gao H, Jiang W, Liu J, Hao G-Z, Xiao L, Ke X, Chen T (2017) Synthesis and characterization of a new co-crystal explosive with high energy and good sensitivity. J Energ Mater 35:490–498
Ding X, Gou R-J, Ren F-D, Liu F, Zhang S-H, Gao H-F (2016) Molecular dynamics simulation and density functional theory insight into the cocrystal explosive of hexaazaisowurtzitane/nitroguanidine. Int J Quantum Chem 116:88–96
Gao H-F, Zhang S-H, Ren F-D, Liu F, Gou R-J, Ding X (2015) Theoretical insight into the co-crystal explosive of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20)/1,1-diamino-2,2-dinitroethylene (FOX-7). Comput Mater Sci 107:33–41
Feng R-Z, Zhang S-H, Ren F-D, Gou R-J, Gao L (2016) Theoretical insight into the binding energy and detonation performance of ε-, γ-, β-CL-20 cocrystals with β-HMX, FOX-7, and DMF in different molar ratios, as well as electrostatic potential. J Mol Model 22:1–14
Wei X-F, Zhang A-B, Ma Y, Xue X-G, Zhou J-H, Zhu Y-Q, Zhang C-Y (2015) Toward low-sensitive and high-energetic cocrystal III: thermodynamics of energetic–energetic cocrystal formation. Cryst Eng Comm 17:9037–9047
Gao H-F, Zhang S-H, Ren F-D, Gou R-J, Wu C-L (2016) Theoretical insight into the temperature-dependent acetonitrile (ACN) solvent effect on the diacetone diperoxide (DADP)/1,3,5-tribromo-2,4,6-trinitrobenzene (TBTNB) cocrystallization. Comput Mater Sci 121:232–239
Han G, Gou R-J, Ren F-D, Zhang S-H, Zhu S-F (2017) Theoretical investigation into the influence of molar ratio on binding energy, mechanical property and detonation performance of 1,3,5,7-tetranitro 1,3,5,7-tetrazacyclo octane (HMX)/1-methyl-4,5-dinitroimidazole (MDNI) cocrystal explosive. Comput Theor Chem 1109:27–35
Ravi P, Badgujar DM, Gore GM, Tewari SP, Sikder AK (2011) Review on melt cast explosive. Prop Explos Pyrotech 36:393–403
Nielsen AT, Christian SL, Moore DW, Nadler MP, Nissan RA, Vanderah DJ, Gilardi RD, George CF, Flippen-Anderson JL, Chafin AP (1998) Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 54:11793–11812
Chen L-Z, Song L, Cao D-L, Wang J-L (2016) Crystal structure of 3,4-dinitropyrazole, C3H2N4O4. Z Kristallogr—New Cryst Struct 231:1099–1100
Sun H (1998) COMPASS: an ab initio force-field optimized for condensed-phase applications—overview with details on alkane and benzene compounds. J Phys Chem B 102:7338–7364
Wei C-X, Huang H, Duan X-H, Pei C-H (2011) Structures and properties prediction of HMX/TATB co-crystal. Propell Explos Pyrot 36:416–423
Li Y-X, Chen S-S, Ren F-D (2015) Theoretical insights into the structures and mechanical properties of HMX/NQ cocrystal explosives and their complexes, and the influence of molecular ratios on their bonding energies. J Mol Model 21:245
Andersen HC (1980) Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384–2393
Allan DR, Clark SJ, Brugmans MJP, Ackland GJ, Vos WL (1998) Structure of crystalline methanol at high pressure. Phys Rev B 58:R11809
Accelrys Software Inc. (2013) Materials Studio release notes, release 7.0. Accelrys Software Inc., San Diego
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chim Acta 120:215–241
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem 27:1787–1799
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, revision A.01. Gaussian, Inc., Wallingford
Lu T, Chen FW (2012) Multiwfn: A multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Bondi A (1964) van der Waals volumes and radii. J Phys Chem 68:441–451
Razas I (2007) On the nature of hydrogen bonds: an overview on computational studies and a word about patterns. Phys Chem Chem Phys 9:2782–2790
Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ (2011) Definition of the hydrogen bond (IUPAC recommendations 2011). Pure Appl Chem 83:1637–1641
Barckholtz C, Barckholtz TA, Hadad CM (1999) C−H and N−H bond dissociation energies of small aromatic hydrocarbons. J Am Chem Soc 121:491–500
Lu T, Chen F-W (2013) Bond order analysis based on the Laplacian of electron density in fuzzy overlap space. J Phys Chem A 117:3100–3108
Rice BM, Hare JJ (2002) A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A 106:1770–1783
Lu T, Chen F-W (2012) Atomic dipole moment corrected Hirshfeld population method. Theor Comput Chem 11:163–183
Steiner T (2002) The hydrogen bond in the solid state. Angew Chem Int Ed 41:48–76
Johnson ER, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohen AJ, Yang W-T (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2010) A possible crystal volume factor in the impact sensitivities of some energetic compounds. J Mol Model 16:895–901
Politzer P, Murray JS (2016) High performance, low sensitivity: conflicting or compatible? Propell Explos Pyrot 41:414–425
Lin H, Zhu S-G, Zhang L, Peng X-H, Chen P-Y, Li H-Z (2013) Intermolecular interactions, thermodynamic properties, crystal structure, and detonation performance of HMX/NTO cocrystal explosive. Int J Quantum Chem 113:1591–1599
Landenberger KB, Matzger AJ (2010) Cocrystal engineering of a prototype energetic material: supramolecular chemistry of 2,4,6-trinitrotoluene. Cryst Growth Des 10:5341–5347
Politzer P, Murray JS (2015) Some molecular/crystalline factors that affect the sensitivities of energetic materials: molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J Mol Model 21:25–35
Politzer P, Murray JS (2014) Detonation performance and sensitivity: a quest for balance. Adv Quantum Chem 69:1–30
Pospíšil M, Vávra P, Concha MC, Murray JS, Politzer P (2011) Sensitivity and the available free space per molecule in the unit cell. J Mol Model 17:2569–2574
Politzer P, Murray JS (2014) Impact sensitivity and crystal lattice compressibility/free space. J Mol Model 20:2223–2230
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Wu J-T, Zhang J-G, Li T, Li Z-M, Zhang T-L (2015) A novel cocrystal explosive NTO/TZTN with good comprehensive properties. RSC Adv 5:28354–28359
Wei Y-J, Ren F-D, Shi W-J, Zhao Q (2016) Theoretical insight into the influences of molecular ratios on stabilities and mechanical properties, solvent effect of HMX/FOX-7 cocrystal explosive. J Energ Mater 34:426–439
Sućeska M (2013) EXPLO5 6.0.1. Brodarski Institute, Zagreb
Keshavarz MH, Motamedoshariati H, Moghayadnia R, Nazari HR, Azarniamehraban J (2009) A new computer code to evaluate detonation performance of high explosives and their thermochemical properties, part I. J Hazard Mater 172:1218–1228
Politzer P, Murray JS (2015) Impact sensitivity and the maximum heat of detonation. J Mol Model 21:262–272
Acknowledgments
We gratefully acknowledge the National Key Laboratory of Applied Physics and Chemistry for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 1458 kb)
Rights and permissions
About this article

Cite this article
Zhu, Sf., Zhang, Sh., Gou, Rj. et al. Theoretical investigation of the effects of the molar ratio and solvent on the formation of the pyrazole–nitroamine cocrystal explosive 3,4-dinitropyrazole (DNP)/2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20). J Mol Model 23, 353 (2017). https://doi.org/10.1007/s00894-017-3516-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3516-4