
Abstract
In recent years, techniques involving the use of organisms to remove or neutralize pollutants from contaminated sites have attracted great attention. The aim of bioremediation is to use naturally occurring organisms to degrade dangerous substances to less toxic or non toxic molecules. The gram-negative bacterium Pandoraea pnomenusa strain B-356 (Pp) has been found to be able to transform a persistent class of organic pollutant compounds, namely the biphenyl and polychlorinated biphenyls (PCBs). A key enzyme in the PCB catabolic pathway is NAD-dependent cis-2,3-dihydrobiphenyl-2,3-diol dehydrogenase (BphB), for which the crystal structure from Pp has been crystallized in apo-, NAD-bound and biphenyldiol-/NAD-bound forms. The substrate binding loop structure has not been completely resolved to date in the former two bound states. Here we report the results of the first extensive molecular dynamics simulations on the three different states of PpBphB. This allowed an in depth characterization of the mechanism of ligand uptake and binding, including unraveling of the gating mechanism. Our simulations give a deep insight into several dynamic features of the enzyme that were not captured by crystal structures.

Starting geometries (in grey) and representative structures of the substrate binding loop of the most populated cluster of structures found in the apo (left), binary(center) and holo (right) forms of PpBphB
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
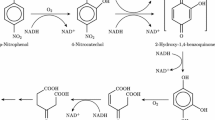
Biphenyl/polychlorinated biphenyls (PCBs) are serious environmental pollutants that threaten both the natural ecosystem and human health [1]. Several approaches that exploit the potential of microbes to degrade PCBs have been developed for remediation of environments contaminated with PCBs [2]. In this regard, Pandoraea pnomenusa strain B-356 (hereafter Pp) has been reported to be more potent towards the persistent congeners 2,6-dichlorobiphenyl and 2,4-dichlorobiphenyl than the other PCB degraders [3]. Due to its specific characteristics, Pp might be exploited effectively in bioremediation protocols against PCB contamination. Understanding the molecular basis of ligand–target interactions in this system may be crucial for a complete characterization of the mechanism of action of the present strain and for future applications in bioremediation protocols of PCB-contaminated sites. In this work we focused particular attention on NAD-dependent cis-2,3-dihydrobiphenyl-2,3-diol dehydrogenase (EC 1.3.1.56; BphB) from Pp. PbBphB is a member of the short-chain dehydrogenase/reductase (SRD) family [4, 5] and catalyzes the second step of the PCB catabolic pathway in bacteria [6]. In its functional form, PpBphB is a homo-dimer composed of two 29.4-kDa monomers. To gain a deeper insight into the reaction mechanism of PpBphB, Dhindwal and collaborators [7] solved the crystal structures of the protein in three forms: the apo-bound form, the coenzyme-NAD (NAD)-bound form (or binary form) and the ternary complex (hereafter holo form), which comprises NAD and the reaction product (2,3-dihydroxybiphenyl, BPY) (Fig. 1).

a Structural alignment of the holo, binary and apo forms of PpBphB dimers (PDBid: 3ZV5, 2Y99, and 2Y93, respectively). The proteins are oriented in order to show the dimerization interface. In the left chain the holo, binary and apo forms of the proteins are colored in dark gray, light gray and white, with the substrate binding loop in dark red, orange and yellow, respectively. The missing parts of the substrate binding loop are indicated by dashed lines. In the right monomer, the proteins are colored dark blue to dark green to correspond with the N- and C-terminals, respectively. BPY and NAD are shown in ball-and-stick format, colored accordingly to atom type. b Detail of the binding site as found in the crystal structure of the holo form. The substrate binding loop has been removed for clarity. BPY and NAD are shown in ball-and-stick format, while the tetrad of residues involved in the reaction are shown as sticks. Atoms are colored accordingly to atom type
Each PpBphB monomer shows a core unit formed by seven parallel β strands [residues 7–11 (β1), 31–36 (β2), 53–57 (β3), 82–84 (β4), 136–140 (β5), 178–184 (β6), and 248–251 (β7)] surrounded by six α-helices, three on each side [residues 15–28 (α1), 39–48 (α2) and 63–77 (α3), 115–133 (α4), 153–173 (α5), and 230–236 (α6)] [8]. The pocket between strand β6 and helix α8 is the substrate-binding region and corresponds to a disordered region (Leu199–Ser206) in the structures of the apo and binary forms. The C-terminal region consists of a loop and a small helix oriented next to helices α5 and α6 of the vicinal molecule in the asymmetric unit. In the ternary form, the loop next to the binding cavity (composed by residues 190–220; hereafter referred to as the substrate binding loop) [7] comes close to the active site and forms a compact cavity where the product can bind. A tetrad of residues (Asn115, Ser142, Tyr155, and Lys159) and the nicotinamide ring of NAD form the active site of the enzyme. In the holo form, the substrate binding loop creates a cavity able to accommodate the BPY ligand. Ser142 is involved in the interaction with the substrate by forming two hydrogen bonds with BPY, one with its 2-OH group and the other with its 3-OH group. Asn143 makes an H-bond with the 2-OH of biphenyl-diol. The hydroxyl group of Tyr155 interacts with the 3-OH of the ligand. The hydroxyl group of the ring directed toward NAD forms H-bonds with Tyr155 and Ser142 (Fig. 1b).
From analysis of the crystal structures, a ligand binding mechanism involving a conformational change of the substrate binding loop was proposed [7]. Moreover, the conformational change of the substrate binding loop leads to the complete formation of a binding cavity, and this was proposed to happen only if both ligands (NAD and BPY) are present at the same time. Indeed, while the latter authors were able to resolve the coordinates of the entire substrate binding loop in the structure of the ternary complex, they were not able to do so in either the apo or the binary forms. In facts, these structures lack seven residues (from 199 to 206) due to insufficient interpretable electron density [7] (Fig. 1). Despite all the above information, a detailed characterization of the process is still missing. Understanding the molecular basis of ligand–target interactions in PCB degradation pathways is crucial to the complete characterization of the mechanism of action of the enzyme and for future applications in bioremediation protocols. The dynamic properties of molecular systems are sometimes not easily accessible by experimental methods. Here, we present three extensive molecular dynamics (MD) simulations starting from the crystal structure of the PpBphB ternary complex. In the first case we simulated the holo form; in the second simulation we started from the ternary complex structure depleted of the BPY ligand to mimic the binary form; and finally, in the third case, we studied the ternary complex depleted of both ligands in order to reproduce the apo form. Our simulations suggest that the presence of NAD molecules is able to induce a local conformational change in the binding cavity, facilitating BPY binding. On the other hand, the presence of both ligands in the binding cavity stabilizes the substrate binding loop in a specific conformation that is favorable for enzymatic activity.
Materials and methods
Structure preparation
We started from the crystal structure of the holo form of PpBphB (PDB ID: 3ZV5) [7]. The holo form was obtained by removing the planar BPY ligand and re-inserting it using a docking protocol (see below). The binary form was obtained by removing the BPY ligand and the apo form was simulated by removal of both ligands. We used the holo structure depleted of one or both ligands because the apo and binary form structures lack the substrate binding loop and because the protein scaffold is essentially the same in all three forms (see Table S2 in the Supplementary Information). The missing residues in the substrate binding loop of chain B of the holo form were modelled by duplicating the coordinates from chain A. Histidine protonation was assessed using the program UCSF Chimera [6].
Docking calculations
The HADDOCK 2.1 (high ambiguity driven biomolecular docking) web server [9] was used to insert the BPY in twisted rings conformation into the binding site of the ternary holo form (the twisted ring conformation was chosen after DFT calculation, see Supplementary Information). The twisted conformation was obtained by constraining the two aromatic rings with a torsion angle of 40°. In the first HADDOCK docking round, a rigid body energy minimization was carried out, and 1,000 structures were calculated. The 200 best solutions selected based on the intermolecular energy were used for the semi-flexible, simulated annealing followed by an explicit water refinement. The solutions were clustered using a cut-off of 7.5 Å root mean square deviation (RMSD) based on the pair-wise backbone RMSD matrix. The best complex was selected based on the HADDOCK score [9] among those in the most populated cluster of structures (see Fig. S2).
Molecular dynamics simulations
The three systems described previously were placed in a water box (7.0 × 7.3 × 6.3 nm each) using a 1.0 nm buffer zone of solvent around the proteins. The resulting systems consisted of ca. 54,000 atoms. The Gromos96 43a1 force field [10] (for the protein) and the SPC/E water model [11] were used. Parameters for BPY and NAD were derived using the PRODRG server [12]. The systems were neutralized by adding Na+ and Cl− ions using the genion program of the GROMACS 4.5.5 package [13]. Each system was energy-minimized and then equilibrated at 300 K and 1 atm by performing 1 ns of gradual annealing using GROMACS 4.5.5 [13]. Each system was geometry optimized in four cycles. In the first two cycles, comprising 800 steps of steepest descent followed by 3,000 steps of conjugate gradient, water molecules were relaxed while the protein was constrained using a harmonic potential with a force constant of 1,000 J mol−1 nm−2. In the third and in fourth cycles, the procedure was repeated without applying any constraint. During the equilibration phase, positional constraints were applied on the protein atoms and on the ligands (force constant of 1,000 J mol−1 nm−2). The temperature and pressure were controlled using a Berendsen thermostat and barostat [14], respectively. An integration step of 2 fs was used and the structures were sampled every 0.1 ps. The LINCS algorithm [15] was used to constrain all bond lengths involving hydrogen atoms. Periodic boundary conditions (PBC) were applied. The particle mesh Ewald (PME) method [16] was used to calculate electrostatic interactions. The cut-off values for the real part of the electrostatic interactions and for the van der Waals interactions were set to 10 Å. For the production runs, a Nosè-Hoover thermostat [17, 18] at 300 K and an Andersen-Parrinello-Rahman Barostat [19, 20] at 1 atm were used. Clustering analysis was performed with the g_cluster module of Gromacs, using the Gromos algorithm [21]. A 0.15 nm cut-off for the RMSD was used to include structures in the same cluster (see SI). The analysis of structural features was carried out on representative structures of the most populated cluster of structures.
Results and discussion
BPY conformational analysis and initial binding pose
In the crystal structures reported by Dhindwal and collaborators [7], the distal rings of the BPY in the ternary form were not very well defined (as stated by the authors) in the electron density map, and presented a planar conformation. However, the structure of BPY present in the PubChem data base (entry CID 254) [22] is a twisted rings conformation. In order to determine the correct BPY conformation to use in our MD simulations, and to estimate the energy barrier needed to switch between the planar and the twisted conformations of BPY, we performed high level ab initio calculations starting from the BPY structure found in the holo form of the protein. The result of quantum calculations (reported in Fig. S1) showed that the twisted form of BPY is more stable with respect to the planar form, which expressed a conformational transition state by 17.6 kJ mol−1. It is thus very unlikely that the planar conformation reported in PpBphB holo crystal structure is the minimal energy BPY conformation in the binding pocket. The twisted form of BPY was then inserted in the binding pocket using virtual docking experiments in order to obtain a new starting configuration for the holo system.Footnote 1 These docking calculations were conducted using the program HADDOCK, which includes a final refinement step with MD in explicit water that allows for flexibility of the ligand and of specified residues. This feature has the positive side effect of including side-chain optimization of the binding cavity [23, 24]. The resulting BPY binding pose is in excellent agreement with the experimental electron density of holo PpBphB (Fig. S3).
Dynamic behavior of the substrate binding loop
To gain a deeper understanding of the structural and dynamic behavior of the binding site of PpBphB, a 200 ns MD simulation was run on each of the three systems described above, totalizing 0.6 μs total simulation time. The RMSD of the backbone atoms stabilized to ca. 0.35–0.40 nm after about 40 ns of simulation time for the apo and binary forms, and after about 130 ns for the holo form (Fig. 2), indicating that the simulations was long enough to relax and equilibrate the molecules. We used the last 70 ns of each simulation for the analysis.

Root mean square deviation (RMSD) vs time plot for simulations performed on the apo form, binary system and holo form. Grey lines represent the effective sampling of the RMSD during the simulation, black lines were obtained by applying a fast Fourier transform filter to reduce noise
The holo form initially explored a conformation at RMSD ca. 2.5 nm (70 % of simulation time) and then relaxed in its final conformation (30 % of simulation time). A cluster analysis (Fig. S6) performed on the trajectory of the holo form, and a structural comparison performed using the LGA structural alignment server [25] revealed that the initial conformation is characterized by a different conformation of helix α4 and the C-terminal region of the protein (Fig. S7). Both of these regions are located far away from the ligand binding pocket. The RMSD values were calculated by LGA server, excluding the C-terminal region, and equilibrated to RMSD values of about 2.6 nm. The high mobility of the C-terminal region was also confirmed by analysis of the root mean square fluctuation (RMSF, Fig. S9) calculated over the last 70 ns of the simulations.
The RMSF values of the substrate binding loop (Fig. 3a) were lower for the holo form with respect to the binary and apo forms (average RMSF of residues 199–206 in chain A/B = 0.17, 0.19, and 0.22 nm, respectively, see Table S1). This suggests that, in the holo form, the substrate binding loop reaches a more stable conformation, probably due to the creation of interactions with BPY and/or NAD, while in the other forms, that region of the binding loop is more mobile and less constrained (Fig. 3b–d).

a Average root mean square fluctuation (RMSF) values for the substrate binding loop calculated using the last 70 ns of the simulations. b–d Details of the substrate binding loop and comparison between the starting geometry (grey) and the representative structure of the most populated cluster of structures found in the apo (b), binary (c), and holo (d) forms of PpBphB
In order to explain this trend in the mobility of the substrate binding loop, we next analyzed the catalytic pocket of the holo form (Fig. 4), focusing primarily on residues in the proximity of BPY and NAD that are considered essential for the catalytic function of the enzyme [1, 7]. Dhindwal et al. [7] found Ser142, Asn143, Tyr155 and Lys159 as the catalytic tetrad that, together with the NAD cofactor, forms the active site. Previous studies suggested that, in the SDR protein family, the conserved Tyr155 residue acts as the catalytic base [26], whereas Ser142 stabilizes the substrate and Lys159 interacts with the nicotinamide ribose of NAD and lowers the pKa of the Tyr155 OH group. Indeed, Ser142 holds the product via the formation of two H-bonds with BPY: one with its 2-OH group [Ser142(Oγ)-BPY(O2)] and the other with its 3-OH group [Ser142(Oγ)-BPY(O3)]. The side chain of Asn143 makes a H-bond with the 2-OH group of BPY [Asn143(Nδ2)-BPY(O2) and Asn143(Oδ1)-BPY(O2)]. Finally, the hydroxyl group of Tyr155 interacts with the 3-OH group of BPY [Tyr155(Oη)-BPY(O2)] [7].

Detail of the holo PpBphB binding site. Lys159, Tyr155, Ser142, and Asn143 are represented using violet, black, red and green sticks, respectively. The NAD and the BPY ligands are reported as orange sticks and cyan balls and sticks, respectively. The substrate binding loop is highlighted in blue, while the hydrophobic residues in contact with the BPY ligand are in yellow. Blue dotted lines Hydrogen bonds
In the last 70 ns of the PpBphB holo simulation, Asn143 forms a H-bond with BPY, while Lys159 forms a H-bond with NAD for 32 % and 31 % of the simulation time, respectively (see Figs. S10, S11). Ser142 and Tyr155 contribute to stabilizing the pose of BPY by forming H-bonds for 6 % and 2 % of the simulation time, respectively. Finally, Tyr155 also forms a H-bond with NAD for 10 % of the simulation time. Similar interactions between Tyr155 and Lys159 are observed in the simulation of the binary form. In particular, NAD orients the conformation of the Tyr155 side chain, which in turn is in a favorable orientation to form a H-bond with Ser142, in preparation for binding of BPY. Stabilization of this interaction is also evident in the holo form, in which it is maintained for about 80 % of simulation time (Fig. S12). Simulation in the apo form confirms this, where the distance between Ser142 and Tyr155 is highly variable (Fig. S12). These observations give us a deeper insight into the specific interactions formed by the residues in the substrate binding loop. Indeed, in the holo form, BPY stabilizes the conformation of the binding loop by interacting through the aromatic rings of BPY with Val207 and Pro208 residues (Fig. 4). Furthermore, as reported in ref. [7], Ile204 makes a hydrophobic interaction with the NAD cofactor only in the holo simulation. The lack of the latter interaction in the apo and in the binary forms explains the extended mobility of this region in the other two binding states of the protein.
Conclusions
Understanding the molecular basis of ligand-target interactions in the PCB degradation pathway is fundamental to a complete characterization of the mechanism of action of the bacterial strain under investigation in the present work and for future applications in bioremediation protocols of contaminated sites. However, investigations into the dynamic properties of molecular systems are not easy using experimental methods. For this reason, performing extensive MD simulations allows us to glean important information in this regard. In the present study, we collected a large amount of data on the dynamic properties of PpBphB, with particular reference to the substrate binding region of the enzyme. In particular, combining the analyses performed on the binding cavity, the studies on the loop stabilization, and the correlated motion calculations for the three systems along the simulations (see Fig. S8), we were able to highlight the dynamic characteristics of the substrate binding loop that determine gating of the enzyme. Indeed, in these simulations, the substrate binding loop in the apo and in binary forms was more mobile with respect to the corresponding holo form, which appears much more stable. It seems clear from our simulations that BPY stabilizes the substrate binding loop in a specific conformation close to the rest of the protein. This conformation is prepared by the NAD, which stabilizes the side chain of Tyr155 and makes a H-bond with Ser142, thus providing the optimal positioning of the sidechains that will receive BPY, by allowing the creation of a network of hydrophobic interactions that contribute to the stabilization of the entire binding pocket. Our MD simulations show that the correlated motions of the three states map to a very similar low frequency motion. The latter indicates that the gating mechanism of the enzyme involves only such conformational changes of the substrate binding loop that allow entry of the ligand upon interaction with the cofactor, in agreement with the three-states crystal structures.
Notes
Preliminary MD calculations done using the BPY binding pose found in the crystal structure showed that the ligand was not stable inside the binding cavity (data not shown).
References
Furukawa K (2003) Trends Biotechnol 21:187–190
Ohtsubo Y, Kudo T, Tsuda M, Nagata Y (2004) Appl Microbiol Biotechnol 65:250–258
Gómez-Gil L, Kumar P, Barriault D, Bolin JT, Sylvestre M, Eltis LD (2007) J Bacteriol 189:5705–5715
Kallberg Y, Oppermann U, Jörnvall H, Persson B (2002) Eur J Biochem 269:4409–4417
Sylvestre M, Hurtubise Y, Barriault D, Bergeron J, Ahmad D (1996) Appl Environ Microbiol 62:2710–2715
Fukuda M (1994) Bioprocess Technol 19:821–835
Dhindwal S, Patil DN, Mohammadi M, Sylvestre M, Tomar S, Kumar P (2011) J Biol Chem 286:37011–37022
Patil DN, Tomar S, Sylvestre M, Kumar P (2010) Acta Crystallogr Sect F: Struct Biol Cryst Commun 66:1517–1520
de Vries SJ, van Dijk M, Bonvin AM (2010) Nat Protoc 5:883–897
van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG (1996) Biomolecular simulation: the gromos96 manual and userguide. Hochschuleverlag AG an der ETH Zürich
Kusalik PG, Svishchev IM (1994) Science 265:1219–1221
Schuttelkopf AW, van Aalten DM (2004) Acta Crystallogr D Biol Crystallogr 60:1355–1363
Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) Bioinformatics 29:845–854
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) J Chem Phys 81:3684–3690
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) J Comput Chem 18:1463–1472
York DM, Wlodawer A, Pedersen LG, Darden TA (1994) Proc Natl Acad Sci USA 91:8715–8718
Hoover WG (1985) Phys Rev A 31:1695–1697
Nosé S (2002) Mol Phys 100:191–198
Nosé S, Klein ML (1983) Mol Phys 50:1055–1076
Parrinello M, Rahman A (1981) J Appl Phys 52:7182–7190
Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE (1999) Angew Chem Int Ed 38:236–240
Bolton EE, Wang Y, Thiessen PA, Bryant SH (2008) PubChem: integrated platform of small molecules and biological activities. In: Ralph AW, David CS (eds) Annual Reports in Computational Chemistry. Elsevier, Amsterdam, pp 217–241
Marchiori A, Capece L, Giorgetti A, Gasparini P, Behrens M, Carloni P, Meyerhof W (2013) PLoS One 8:e64675
Sandal M, Duy TP, Cona M, Zung H, Carloni P, Musiani F, Giorgetti A (2013) PLoS One 8(9):e74092
Zemla A (2003) Nucleic Acids Res 31:3370–3374
Kavanagh KL, Jörnvall H, Persson B, Oppermann U (2008) Cell Mol Life Sci 65:3895–3906
Acknowledgments
F.M. was funded by CIRMMP (Consorzio Interuniversitario di Risonanze Magnetiche di Metallo-Proteine).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1630 kb)
Rights and permissions
About this article

Cite this article
Piccoli, S., Musiani, F. & Giorgetti, A. Dynamic characterization and substrate binding of cis-2,3-dihydrobiphenyl-2,3-diol dehydrogenase—an enzyme used in bioremediation. J Mol Model 20, 2531 (2014). https://doi.org/10.1007/s00894-014-2531-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2531-y