
Abstract
Dye-sensitized solar cells (DSSCs) have drawn great attention as low cost and high performance alternatives to conventional photovoltaic devices. The molecular design presented in this work is based on the use of pyran type dyes as donor based on frontier molecular orbitals (FMO) and theoretical UV-visible spectra in combination with squaraine type dyes as an acceptor. Density functional theory has been used to investigate several derivatives of pyran type dyes for a better dye design based on optimization of absorption, regeneration, and recombination processes in gas phase. The frontier molecular orbital (FMO) of the HOMO and LUMO energy levels plays an important role in the efficiency of DSSCs. These energies contribute to the generation of exciton, charge transfer, dissociation and exciton recombination. The computations of the geometries and electronic structures for the predicted dyes were performed using the B3LYP/6–31+G** level of theory. The FMO energies (EHOMO, ELUMO) of the studied dyes are calculated and analyzed in the terms of the UV- visible absorption spectra, which have been examined using time-dependent density functional theory (TD-DFT) techniques. This study examined absorption properties of pyran based on theoretical UV- visible absorption spectra, with comparisons between TD-DFT using B3LYP, PBE, and TPSSH functionals with 6–31+G (d) and 6–311++G** basis sets. The results provide a valuable guide for the design of donor-acceptor (D-A) dyes with high molar absorptivity and current conversion in DSSCs. The theoretical results indicated 4-(dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran dye (D2-Me) can be effectively used as a donor dye for DSSCs. This dye has a low energy gap by itself and a high energy gap with squaraine acceptor type dye, the design that reduces the recombination and improves the photocurrent generation in solar cell.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The high performance and low cost of DSSCs have drawn great interest from both academic and industrial circles. The improvement of solar energy-to-electricity conversion efficiency has continued to be an important research area of DSSCs [1–10]. The performance of DSSCs firstly depends on the relative HOMO–LUMO energy levels of the sensitizer. Therefore, efficient electron injection requires the LUMO level of the dye to be higher in energy than the TiO2 conduction band edge, whereas the HOMO of the dye level may lie below the energy level of the electrolyte to allow efficient regeneration of the oxidized dye. The electronic structures, such as FMO (EHOMO, ELUMO, and Eg = ELUMO-EHOMO) of dye molecules in DSSC are deeply related to the electron transfer by photoexcitation. In literature there have been many studies to design and improve the efficiency of organic dyes [11–18].
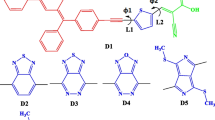
DFT calculations were employed to analyze the electronic structure of organic dyes which are used in DSSC [19–28]. In this paper, we try to design organic dyes based on pyran as donor in their charge-transfer chromophoric system. The theoretical and experimental studies of pyran dyes have been extensively investigated [29–32]. These dyes are used in many applications such as bulk-heterojunction solar cells, organic light emitting diode (OLED) applications, and sensors [33–37]. The chemical structure of these dyes (Scheme 1) consists of a donor moiety (arylamine moiety) and an acceptor moiety (dicyanovinyl moiety) connected by a π-conjugated structure. The band gap energy (Eg), which is the energy gap between LUMO and HOMO in the donor itself or between the HOMO in the donor and the LUMO in the acceptor dye, is a key factor which determines the efficiency of donor-acceptor system in solar cell. Thus, control of this energy gap by structural modification is extremely important in the design of low Eg dyes. The main aim of this work is to provide new electronic properties for designing efficient donor dyes. We employed TD-DFT [38–41] calculations to compute the excitation spectrum.

The molecular structures of the studied organic dyes in the present work
Computational methods
Calculations were performed in ground state using Gaussian09 [42]. The studied molecules were optimized using B3LYP/6–31+G** [43–46] level of theory calculations. Frequency calculations were carried out at the same levels of the theory in order to characterize the stationary points as local minima. TD-DFT theory is presently enjoying enormous popularity in quantum chemistry, as a useful tool for extracting electronic excited state energies. Based upon the optimized geometry, the absorption properties in gas phase were investigated using TD-DFT at the same level of the theory. The molecular orbital structures and energies were also calculated at the B3LYP method with 6–31+G** basis sets for both HOMO and LUMO levels. UV-absorption and molecular orbital energies preformed with different functionals, including the hybrid functionals PBE [47], and TPSSH [48–50] with high basis set 6–311++G**. The basic molecular structures of this study are shown in Scheme 1. These dyes are designed to investigate the effect of different donating and withdrawing groups on geometrical and electronic properties of such donor dyes with the objective to improve the efficiency of photocurrent dyes in DSSCs. Also, these pyran type dyes have been optimized and investigated using the DFT.
Results and discussion
Geometrical and conformational analysis
The free rotation of the parent compound about single bonds between the rings has been investigated so as to find the most stable conformers before analyzing the substituent effect on the geometrical parameters and electronic structure. Since the parent compound (D) can rotate about C7-C8 and C5-C6 single bonds, therefore, we carried out conformational analysis using B3LYP/6–31+G** to provide the most stable conformers for the studied molecules. As the rotational process of the two single bonds are the same, we are concerned with only one of them. The potential energy surface (PES) scan for the parent molecule is shown in Fig. 1; all the geometrical parameters were simultaneously relaxed while the dihedral angles C10-C8-C7-C6 are varied in steps of 20° ranging from 0° to 200°. For the rotation of phenyl ring (C10-C8-C7-C6), there are two minimal energy points on PES (180° and 00° which point to the most stable conformers. The stationary points were confirmed by the frequency analysis as minima with all real frequencies. The calculations indicate that the transition states have one imaginary frequency. The transition state of the rotation is found for a torsion angle at 100° with a barrier of about 8.0 kcal mol−1, respectively. The planar conformer that has dihedral angle 0.0° is less stable than the most stable planar conformer that has dihedral angle 180° by 2.0 kcal mol−1. Thus, it is evident that the planar conformer is the most stable form. Therefore, in the present work we have focused on this form of the parent molecule to clarify its molecular structure, electronic properties, and UV- visible spectra.

Potential energy surface (PES) scan of the calculated energies vs C10-C8-C7-C6 dihedral angle of the parent compound (D) using B3LYP/6–31+G** level of theory. (Relative energies are in kcal mol−1 relative to the most stable conformer at 180°)
The optimized geometries of the studied pyran dyes D1, D2, D3, D4, D1-Me, D2-Me, D3-Me, D4-Me, A1, and A2 are shown in Figs. 2, 1S, and 3. The selected geometrical parameters, which include bond lengths, and charge density distribution, are summarized in Figs. 2 and 3. The calculated geometrical data indicate that the geometrical parameters for all donor dyes are very similar. It is observed that the mean values of the single bonds C7-C8 and C5-C6 distance by B3LYP/6-31+G** method are 1.460 Å and 1.450 Å, respectively, and the value of the double bond C6-C7 distance is 1.351 Å. This can be understood from the localized charge density on whole molecules. We did not observe important changes in the internuclear distances between the studied dyes. The central double bond and single bonds remained constant. Figure 3 displays the optimized structure of the two studied squaraine dyes as acceptor dyes.

Optimized geometries for the studied donor dyes using B3LYP/6–31+G** level of theory

Optimized geometries for the squaraine dyes using B3LYP/6–31+G** level of theory
Frontier molecular orbital
In the design of dye molecules, the HOMO and LUMO levels and their Eg value are among the most important properties dominating the dye performance in DSSCs. The Eg of donor dyes plays a major role in broadening the range of absorption which improves the photocurrent efficiency of the dyes. At the same time, small Eg value in donor dyes aids in the generation of excitons and increases the efficiency of photoexcitation. Eg values in donor and between donor and acceptor dyes are very important for the generation and recombination of charges. The energies of the main molecular orbital, HOMO, and LUMO were calculated using B3LYP/6–31+G** level of theory in gas phase, and the other two density functional (PBEPBE, TPSSH) with 6–311++G** basis set to validate the level of theory. Experimental data for 4-(dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran (D2-Me) are available in the literature [51]. The theoretical Eg was estimated as Eg = ELUMO-EHOMO. Tables 1 and 2 summarize the energies of FMO for the studied dyes at different levels of theory. In this section, the energies of HOMO, LUMO, and gap energies for the studied dyes are calculated. Also an interaction between HOMO present in the donor and LUMO of squaraine acceptor is investigated, as shown in Fig. 4. It is clear that the insertion of methyl group in heterocyclic ring reduces the gap energies (LUMO-HOMO) for all the studied donor dyes. The data in Tables 1 and 2 show a small effect on the HOMO and LUMO levels upon changing the substituent groups. The gap energies of D and D-Me are 3.322 and 3.207 eV using B3LYP/6–31+G** level of theory, respectively. This decrease in Eg is also clear in D3 and D3-Me which is 0.15 eV. The HOMO energy of D2-Me is 5.61 eV, which is in a good agreement with the experimental value (5.56 eV) with error equal to 0.05 eV using B3LYP/6–31+G**. PBEPBE/6–311++G** level of theory shows good behavior for LUMO energy calculation. ELUMO energy is 3.13 eV compared with the experimental value 3.43 eV [51]. The high performance of DSSCs based on organic dyes is probably due to the high open-circuit voltage (Voc) values with large gap energies between donor (pyran) and acceptor (squaraine) dyes (EHOMO (donor) -ELUMO (acceptor)). To investigate this problem, two different squaraine structures as acceptor dyes were investigated. Figure 4 presents the recombination charges process in donor-acceptor system. As shown in this figure, the structure A2 is better than A1 as acceptor dye with the donor under examination. The result suggests that charge recombination does not occur more easily in D2-Me and D3-Me. The gap energy between D3-Me and D2-Me with acceptor dye are 2.33 and 3.32 eV using B3LYP/6–31+G**. This result indicates that charge recombination occurs more easily in D2-Me than in D3-Me. In conclusion, D2-Me dye displays high performance as donor dye which has low gap energies by itself (photocurrent) and with acceptor dye as well.

The electronic absorption spectra
Development of new sensitizers that can absorb in the visible region with relatively large absorption coefficients is also desired in designing organic dyes for DSSCs. TD-DFT//B3LYP/6-31+G** calculations on electronic absorption spectra in gas phase were performed to understand the electronic transitions of the studied dyes in this work. TPSSH, PEBPEB, and B3LYP with 6-311++G** were tested to compare maximum absorption wave lengths. The absorption spectra, excitation energy, oscillator strength, and main configurations are listed in Table 3. TD-DFT results in Tables 3 and 4 include only the first three singlet transitions of absorption bands with the oscillator strength larger than 0.01. Triplet transitions were summarized in Table 4 using B3LYP/6-311++G** level of theory. The wavelength longer than 300 nm was recorded in Table 3 because the absorption in visible and near-UV regions is the most important region for photo-to-current conversion. The present results indicate that the absorption bands of the donor dyes are red shifted under the effect of substituents. The parent dye (D) has maximum absorption band at 418 nm which is the smallest value. The absorption band that corresponds to the maximum absorption of the other substituent donor dyes is red shifted except in NH2 group (D4). The insertion of dimethylamino (D2) causes large red shifts in which the absorption band is 441 nm. Most of the electronic transitions are mainly contributed by the LUMOs-HOMOs transition. All absorption bands in the visible region are typical π–π* transitions. The maximum absorption bands for the studied dyes resulted from the electronic transitions from the initial states that are mainly contributed by the HOMO-1-LUMO are shown in Tables 2. These dyes exhibit strong charge transfer absorption bands in the visible region. The lowest transition of D2-Me and D3-Me dyes is calculated at 2.81 and 2.48 eV (Table 3) and corresponds to the intramolecular charge transfer (CT) excitation from the HOMO (localized on phenyl ring) to the LUMO, localized on hetero pyran ring. As shown in Fig. 2S (supplementary data), the electron distribution in the HOMO and LUMO of all studied derivatives clearly indicates that an intramolecular charge transfer from the donor moiety (phenyl ring) to the acceptor moiety (pyran moiety) occurs upon photoexcitation of the dye (Fig. 5). The results in Tables 2 and 3 were compared with the available experimental data for D2-Me molecule [51]. λmax values calculated from TD-B3LYP, TD-PBE, and TD-TPSSH with 6–311++G** basis set are compared with available experimental data. As shown in Tables 3 and 4, the λmax values calculated using B3LYP with different basis sets 6–31+G** and 6–311++G** are 441.21 and 440.51, respectively. These values are close to experimental value 460 nm. The results in Table 3 indicated that, the best value of λmax for D2-Me was predicted to be 457.92 nm using TPSSH/6–311++G** level of theory which was the closest to experimental value (∼460 nm).

Electron distribution of frontier molecular orbitals of two donor dyes (D3-Me and D2-Me)
The transfer energy between donor and acceptor dyes during absorption and emission spectra is interesting in DSSCs. The plots of electronic absorption spectra of donor derivatives and squaraine acceptor dye are shown in Fig. 6. Figure 6a and b display theoretical absorption overlap between pyran donor and squaraine acceptor dyes. The amount of spectral overlap between the studied donor dyes and acceptor is large in D3-Me and D2-Me. The above mentioned results and the present one confirm and recommend that the structure of D2-Me dye is the best model for DSSC system which can enhance both the spectral breadth and absorption of dye sensitized solar cells.

The absorption spectra of studied compounds obtained by TD-DFT//B3LYP/6–311++G** level of theory. (Green color for acceptor dye and other colors are for donor dyes)
Conclusions
In summary, we performed DFT and TD-DFT calculations to analyze and understand the electronic structure, absorption, and electron transport properties for organic donor dyes. The effect of substituents on donor dye is reflected on the magnitudes of molecular orbital energies, as it is confirmed by the calculated UV–visible spectra and the energies of the FMO. TD-DFT appeared to be an accurate approach. The B3LYP functional with the 6–311++G** basis set produced the most reliable HOMO energy. On other hand, PEBPEB/6–311++G** level of theory is in good agreement with experimental value of ELUMO. TPSSH/6–311++G** level of theory indicated that the value of λmax is 457.92 in gas which agrees well with the experimental value (460 nm). The results indicate that D2-Me showed the appropriate donor dye. It also exhibited the best absorption characteristics in maximum wavelength and high absorption band overlaps with squaraine acceptor dye, as well as performing a good photocurrent character. It can be concluded that the best molecular design for donor dyes based on pyran would be the one which contains dimethylamino group in the para position of phenyl ring and methyl group in pyran ring (D2-Me). A future study is under way for the synthesis and characterization of squaraine and pyran donor-acceptor dyes.
References
Vlachopoulos N, Liska P, Augustynski J, Grätzel M (1988) Very efficient visible light energy harvesting and conversion by spectral sensitization of high surface area polycrystalline titanium dioxide films. J Am Chem Soc 110:1216–1260
O’Regan B, Grätzel M (1991) A low-cost, high-efficiency solar cell based on dye-sensitized colloidal titanium dioxide films. Nature 353:737–740
Smestad G, Bignozzi C, Argazzi R (2004) Testing of dye sensitized TiO2 solar cells I: experimental photocurrent output and conversion efficiencies. Sol Energy Mater Sol Cells 53:259–272
Hagfeldt A, Grätzel M (1995) Light-induced redox reactions in nanocrystalline systems. Chem Rev 95:49–68
Kay A, Grätzel M (1996) Low cost photovoltaic modules based on dye sensitized nanocrystalline titanium dioxide and carbon powder. Sol Energy Mater Sol Cells 44:99–117
Kalyanasundaram K, Grätzel M (1998) Applications of functionalized transition metal complexes in photonic and optoelectronic devices. Coord Chem Rev 177:347–414
Hagfeldt A, Grätzel M (2000) Molecular photovoltaics. Acc Chem Res 33:269–277
Grätzel M (2003) Dye-sensitized solar cells. J Photochem Photobiol C Photochem Rev 4:145–153
Nazeeruddin MK, De Angelis F, Fantacci S, Selloni A, Viscardi G, Liska P, Ito S, Takeru B, Grätzel M (2005) Combined experimental and DFT-TD-DFT computational study of photoelectrochemical cell ruthenium sensitizers. J Am Chem Soc 127:16835–16848
Kafafy H, Wu H, Peng M, Hu H, Yan K, El-Shishtawy R Mm, Zou D (2014) Steric and solvent effect in dye-sensitized solar cells utilizing phenothiazine-based dye. Int J Photoenergydoi:10.1155/2014/548914
Wang ZS, Hara K, Danoh Y, Kasada C, Shinpo A, Suga S, Arakawa H, Sugihara H (2005) Photophysical and (photo) electrochemical properties of a coumarin Dye. J Phys Chem B 109:3907–3914
Wang ZS, Cui Y, Hara K, Dan-oh Y, Kasada C, Shinpo A (2007) A high-light-harvesting-efficiency coumarin dye for stable dye-sensitized solar cells. Adv Mater 19:1138–1141
Alex S, Santhosh U, Das S, Papadodima O, Chatziioannou A, Patrinou-Georgoula M, Kolisis FN, Pletsa V, Guialis A (2005) Dye sensitization of nanocrystalline TiO2: enhanced efficiency of unsymmetrical versus symmetrical squaraine dyes. J Photochem Photobiol A 172:63–71
Burke A, Schmidt-Mende L, Ito S, Grätzel M (2007) A novel blue dye for near-IR “dye-sensitized” solar cell applications. Chem Commun 3:234–236
Howie WH, Claeyssens F, Miura H, Peter LM (2008) Characterization of solid-state dye-sensitized solar cells utilizing high absorption coefficient metal-free organic dyes. J Am Chem Soc 130:1367–1375
Horiuchi T, Miura H, Sumioka K, Uchida S (2004) High efficiency of dye-sensitized solar cells based on metal-free indoline dyes. J Am Chem Soc 126:12218–12219
Campbell WM, Burrell AK, Officer DL, Jolley KW (2004) Porphyrins as light harvesters in the dye-sensitized TiO2 solar cell. Coord Chem Rev 248:1363–1379
Chen Z, Li F, Huang C (2007) Organic D-π-A dyes for dye-sensitized solar cell. Curr Org Chem 11:1241–1258
Walsh PJ, Gordon KC, Officer DL, Campbell WM (2006) A DFT study of the optical properties of substituted Zn(II)TPP complexes. J Mol Struct (THEOCHEM) 759:17–24
Vyas S, Hadad CM, Modarelli DA (2008) A computational study of the ground and excited state structure and absorption spectra of free-base N-confused porphine and free-base N-confused tetraphenylporphyrin. J Phys Chem A 112:6533–6549
Balanay MP, Kim DH (2008) DFT/TD-DFT molecular design of porphyrin analogues for use in dye-sensitized solar cells. Phys Chem Chem Phys 10:5121–5127
Liu T, Zhang HX, Zhou X, Xia BH (2008) Theoretical studies on [Ru(bpy)2(NN)]2+ [NN = hydrazone and azine]: ground- and excited-state geometries, electronic structures, absorptions, and phosphorescence mechanisms. Eur J Inorg Chem 2008:1268–1276
Liu Z (2008) Theoretical studies of natural pigments relevant to dye-sensitized solar cells. J Mol Struct (THEOCHEM) 862:44–48
Balanay MP, Kim DH (2009) Structures and excitation energies of Zn–tetraarylporphyrin analogues: a theoretical study. J Mol Struct (THEOCHEM) 910:20–26
Minaev BF, Baryshnikov GV, Slepets AA (2012) Structure and spectral properties of triphenylamine dye functionalized with 3,4 propylenedioxy thiophene. Opt Spektrosk 112:899–905
Baryshnikov GV, Minaev BF, Myshenko EV, Minaeva VA (2013) Structure and electronic absorption spectra of isotruxene dyes for dye sensitized solar cells: investigation by the DFT, TD-DFT, and QTAIM methods. Opt Spektrosk 115:555–562
Baryshnikova GV, Minaeva BF, Minaeva VA, Ning Z, Zhang Q (2012) Structure and spectral properties of truxene dye S5. Opt Spektrosk 112:193–199
Minaev BF, Gleb VB, Minaeva VA (2011) Electronic structure and spectral properties of the triarylamine-dithienosilole dyes for efficient organic solar cells. Dyes Pigments 92:531–536
Marques MAL, Gross EKU (2004) Time-dependent density-functional theory. Annu Rev Phys Chem 55:427–455
Samuel GA, Jason P, Joshi P, Qiquan Q, Youngjae Y (2004) New pyran dyes for dye-sensitized solar cells. J Photochem Photobiol A Chem 224:116–122
Zhidan T, Yunchang L, Baozhu T, Jinlong Zh (2013) Synthesis and proton-induced fluorescence “OFF–ON” switching of a new D-p-A type pyran dye. Res Chem Intermed
Gerasimenkoa A Yu, Podgaetsky V M, Krasovsky V I, Lugovsky A P (2009) Optical Memory and Neural Networks. (Information Optics) 18:218–222
Cui Y, Jiancan Y, Gao J, Wang Z, Qian G (2009) Synthesis and luminescence behavior of inorganic–organic hybrid materials covalently bound with pyran-containing dyes. Sol gel Sci Technol 52:362–369
Kim JH, Lee H (2002) Synthesis, electrochemistry, and electroluminescence of novel red-emitting poly(p-phenylenevinylene) derivative with 2-pyran-4-ylidenemalononitrile obtained by the heck reaction. Chem Mater 14:2270–2275
Peng Q, Lu Z Y, Huang Y, Xie M G, Han S H, Peng J B, Cao Y (2004) Synthesis and characterization of new red-emitting polyfluorene derivatives containing electron-deficient 2-pyran-4-ylidene-malononitrile moieties, Macromolecules 260–266
Son YA, Gwon SY, Lee SY, Kim SH (2010) Synthesis and property of solvatochromic fluorophore based on D-pi-A molecular system: 2-{[3-Cyano-4-(N-ethyl-N-(2-hydroxyethyl)amino)styryl]-5,5-dimethylfuran-2(5H)-ylidene}malononitrile dye. Spectrochim Acta A Mol Biomol Spectrosc 75:225–229
Xue JLH, Gu X, Yang Z, Xu B, Tian W (2009) Efficient bulk-heterojunction solar cells based on a symmetrical D-pi-A-pi-D organic dye molecule. J Phys Chem C 113:12911–12917
Furche F, Burke K (2005) Time-dependent density functional theory in quantum chemistry. In: Spellmeyer A (ed) Annual reports in computational chemistry, vol 1. Elsevier, Amsterdam, pp 19–30
Burke K, Gross EKU (1998) A guided tour of time-dependent density functional theory. In: Joubert A (ed) Density functionals: theory and applications. Springer, Berlin
Gross EKU, Aobson JF, Petersilka M (1996) Density functional theory of time-dependent phenomena. Top Curr Chem 181:81–172
Luo Y, Jonsson D, Norman P, Ruud K, Vahtras O, Minaev B, Ågre H, Rizzo A, Mikkelsen KV (1998) Some recent developments of high-order response theory. Int J Quant Chem 70:219–239
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Montgomery J A, Jr, Vreven T, Kudin K N, Burant J C (2009) Gaussian 03; Gaussian Inc, Wallingford, CT
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Perdew JP, Burke K, Ernzerhof M (1997) Generalized gradient approximation made simple. Phys Rev Lett 78:1396
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6170
Perdew JP, Tao J, Staroverov VN, Scuseria GEJ (2004) Chem Phys 120:6898–6911
Perdew JP, Kurth S, Zupan A, Blaha P (1999) Phys Rev Lett 82:2544–2547
Wu J, Hagelberg F, Dinadayalane TC, Leszczynska D, Leszczynski J (2011) Do stone–wales defects alter the magnetic and transport properties of single-walled carbon nanotubes? J Phys Chem C 115:22232–22241
Nueesch F, Zuppiroli L, Berner D, Ma C, Wang X, Cao Y, Zhang B (2004) Space charge and polarization effects upon doping organic light-emitting diodes with pyran-containing donor-acceptor molecules. Res Chem Intermed 30:495–507
Acknowledgments
This Project was funded by the King Abdulaziz City for Science and Technology (KACST) under grant number 11-ENE1531-03. The authors, therefore, acknowledge with thanks KACST for support for Scientific Research. Also, the authors appreciate the kind cooperation provided by the Deanship of Scientific Research (DSR), King Abdulaziz University.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
El-Shishtawy, R.M., Asiri, A.M., Aziz, S.G. et al. Molecular design of donor-acceptor dyes for efficient dye-sensitized solar cells I: a DFT study. J Mol Model 20, 2241 (2014). https://doi.org/10.1007/s00894-014-2241-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2241-5