
Abstract
I have performed quantum chemical calculations for the CF3X (X = Cl, Br) ∙∙∙Aun (n = 2, 3, and 4) complexes at M05-2X/aug-cc-pVDZ(PP) level. Two types of optimized structures were obtained. Type I complexes are stabilized by the coordination force between the negative electrostatic potential of halogen atom and the gold atom, and type II complexes contain halogen bonds formed between the σ-hole of the halogen atoms and the negative electrostatic potential of Aun. Results of the interaction energy indicate that type I complexes are more stable than type II complexes. AIM analysis reveals that type II complexes are a closed shell interaction and there is a partially covalent nature for type I complexes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intermolecular interactions play a great role in the fields of chemistry, physics, and biology. In recent years, great progress has been made in the research of different types of intermolecular interactions [1–3]. Halogen bonding is a noncovalent interaction between a halogen-containing molecule and a neutral or anionic molecule, which is similar to hydrogen bonding in terms of directionality and strength [4]. Politzer et al. [5–11] reported that a halogen bond is an interaction between a region of positive potential in the axis of the covalent bond of the halogen atom (called the σ-hole) and a region of negative potential on another molecule.
Halogen-bonded interaction plays a prominent role in crystal engineering, molecular recognition, and biological systems [12–22]. Recently, the supramolecular assembly of gold nanoparticles (AuNPs) mediated by the halogen-bonded interactions has been attracted a deal of attention [23–25]. Van der Boom et al. [23, 24] demonstrated for the first time that the assembly of hybrid organic AuNP structures onto planar functionalized surfaces mediated by halogen bonding. Their findings indicate that the formation of structurally well-defined and task-specific nanoparticle-based assemblies based on halogen bond is a realistic possibility. More interestingly, Blakey and co-workers [25] presented the interaction of iodoperfluorobenzene compounds with AuNPs and well defined halogen-bonded assemblies of AuNPs formed. A question was proposed: what is the nature of the interaction between the halogen atom and AuNPs?
In order to solve this problem, I investigated the X∙∙∙Au interactions in the CF3X (X = Cl, Br) ∙∙∙Aun (n = 2, 3, and 4) complexes with quantum chemical calculations. By studying these complexes, the following questions are answered: 1) Is there a X∙∙∙Au interaction? 2) If this interaction exists, is it a halogen bond or other intermolecular interaction?
Computational details
The geometries of all the complexes were fully optimized by means of the hybrid M05-2X functional [26, 27]. This method has been proven to be reliable for the study of noncovalent complexes [28, 29]. The aug-cc-pVDZ-PP basis set, which uses pseudopotentials to describe the inner core orbitals, was employed for Au, while for other atoms aug-cc-pVDZ was applied. All of the optimized structures were characterized as minima in the potential energy surface by verifying that all the vibrational frequencies are real. The basis set superposition error (BSSE) was eliminated by using the standard counterpoise correction (CP) method of Boys and Bernardi [30]. The above calculations were performed with the Gaussian 09 suite of programs [31].
The quantum theory of atoms in molecules (QTAIM) analysis [32] was performed with the help of AIMAll [33] using the M05-2X wavefunctions generated by Gaussian 09. The electron density shift was calculated by evaluating the difference between the total electron densities of the CF3X∙∙∙Aun complexes and individual moieties (CF3X and Aun), which was fulfilled by the Multiwfn programs [34].
Results and discussion
Geometries and interaction energies
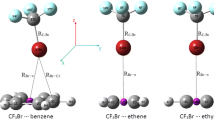
I attempted to search stable structures for the CF3X∙∙∙Aun complexes from different initial configuration. Two types of minimas without imaginary frequencies were obtained. The optimized structures of these complexes are displayed in Fig. 1. For convenience, the left type in Fig. 1 is called type I complex, and the right is type II complex. The most positive MEP, VS,max and the most negative MEP, VS.min in the Br, Cl atoms and the Aun molecules were calculated at the M05-2X level, which are collected in Table 1. I take the surfaces to be the 0.001 electrons/bohr3 contours of the molecular electronic densities, which have been verified to be sufficient to compute MEP by Politzer [5, 8]. The σ-holes in the CF3Cl and CF3Br molecules are in the positive outer regions of the halogen atoms, which are accompanied negative lateral sides. VS,max in the Br atom are larger than that in the Cl atom, which is consistent with previous study [8]. It is interesting that there are negative potentials in Aun, which lies in the plane of the molecules. The molecular surface electrostatic potential of Aun was shown in Fig. 2. One can see from Fig. 1 and 2 that the gold atom interacted with the negative region of the halogen donor in type I complexes, and for type II complexes, the σ-hole along the C−X bond pointed to the plane of Aun. Because the halogen bond was defined as the interaction between the σ-hole and the other molecule, type I complexes do not contain halogen-bonded interaction from the geometrical view. Type I complexes are stabilized by the traditional coordination force [35, 36] between the gold atom and the negative potential of the halogen atom, which has been proven to be essential for the transition metal complex. In type II complexes, the interaction is between the σ-hole of the halogen atom and the negative potential of Aun molecule, which indicates electrostatically-driven halogen-boned interaction.

Optimized geometries of the CF3X (X = Cl, Br) ∙∙∙Aun (n = 2, 3, and 4) complexes

Molecular surface electrostatic potential of Aun (n = 2, 3, and 4), computed on the 0.001 a.u. contour of the electronic density at M05-2X/aug-cc-pVDZ-PP computational level. The blue and red arrows are denoted to be the VS,max and VS,min sites respectively
Table 2 lists the key geometrical parameters and interaction energies for the studied complexes. It is clear that X∙∙∙Au interactions in type I complexes are far away from linear (∠(C−X∙∙∙Au) = 90° ∼ 110°) and the values of ∠(C−X∙∙∙Au) for type II complexes is near to 180°. This again confirms that type I complexes are not halogen bonds because the most essential feature of a halogen bond is the linear interaction between the C−X bond and the negative region of the other molecule. From Table 2 one can see that the distance between halogen and gold atom in type I complexes are smaller than that in type II complexes, which is consistent with the interaction energy. For example, type I complex of CF3Br∙∙∙Au2 are more stabilized than type II complex (9.72 vs 2.71 kcal mol-1), and R (X∙∙∙Au) in the latter is 3.530 Å, larger than the former 2.656 Å. The choice of the gold cluster influences the result slightly. For type I complexes of CF3Br∙∙∙Aun, the interaction energy of CF3Br∙∙∙Au4 exceeds 1.31 kcal mol-1 more than that of CF3Br∙∙∙Au2. The interaction energy of CF3Cl∙∙∙Au3 and CF3Cl∙∙∙Au4 for type I complex is almost the same. In both type I and type II complexes, the Br∙∙∙Au interactions become much stronger in strength than the Cl∙∙∙Au interactions. That is to say, the Br∙∙∙Au coordination force is much stronger than that of Cl∙∙∙Au. The electrostatic potential in the σ-hole of CF3Br is more positive than that of CF3Cl, which decides the stability order of type II complexes. The C−X bonds in all the complexes present a red-shift, which says that the length of the C−X bonds become larger and the vibration frequencies decrease. The degree of the C−X bonds change in type I complexes is larger than that in type II complexes, which is consistent with the previous study [37], indicating that larger interaction energies led to stronger elongation of the C−X bonds.
Electron density shift
In order to obtain a deeper insight into the X∙∙∙Au interaction, we analyzed the electron density shift that accompanied the formation of the complexes. The shifts of electron densities for the CF3Br∙∙∙Aun complexes are illustrated in Fig. 3. It is clearly seen that the shifts of electron density in type I complexes are greater than that in type II complexes. Figure 3 shows that the electron density lost/gain regions are alternated, and there are differences between these two types of complexes. Gold atoms gained large density and fluorine atoms in the CF3Br molecules lost a lot of density in type I complexes. For type II complexes, the region of electron density lost is in the backbone of gold clusters and the region of electron density gain is around the fluorine atoms. Type I and II complexes behaved similarly in the interactive regions, which says that the loss and gain of electron density occurs around the outer regions of CF3Br and Aun molecules respectively.

Shifts of electron density, in a.u., as a result of the CF3Br···Aun complexes. The plots are computed in the 0.001 electrons/bohr3 contours of the molecular electronic densities. Purple regions denote electron gain, and turquoise regions represent electron lost
AIM analysis
AIM theory is based on a topological analysis of the electron charge density and its Laplacian, which has been successfully applied in characterizing hydrogen bonds and halogen bonds of different strengths in a wide variety of molecular complexes. With this in mind, we performed a topological analysis to gain more insights into the nature of the CF3X∙∙∙Aun complexes. The results show that a bond path exists linking the halogen atom with the gold atom accompanied by a bond critical point (BCP) between the two atoms in type I complexes, and a ring critical point (RCP) formed between one halogen atom and several gold atoms is detected in type II complexes. The electron density at the BCPs and RCPs and their Laplacians are summarized in Table 3. For type I complexes, all the electron densities, ρ, are much larger than those of type II complexes, which is in accordance with the interaction energy. One can see a linear relationship between the electron density and the interaction energy from Fig. 4. The linear correlation coefficient for this dependence amounts to 0.912. The Laplacians, ▽2ρ, are positive, indicating the typical noncovalent type of interactions in these complexes.

Relationship between the interaction energy and electron density
The electronic energy density can also provide another insight into the nature of noncovalent interactions. The electronic energy density H, local one-electron kinetic energy density G, and local potential energy density V are also given in Table 3. The sum of G and V is H. G is positive and V is negative. The positive or negative sign of H depends on the relative magnitude of G and V. If the G value is larger than the absolute value of V, the H value is positive, thus a purely closed shell interaction is expected. Otherwise, the negative H indicates that the interactions correspond to some degree of covalent character. From Table 3 one can see that the negative H value is found for type I complexes and the positive H value is obtained for type II complexes. This indicates that type II complexes are a closed shell interaction and there is a partially covalent nature for type I complexes [38].
Conclusions
In summary, I have investigated the noncovalent complexes between CF3X (X = Cl, Br) and Aun (n = 2, 3, and 4) using ab initio calculations. It was found that there were two different optimized structures. One is stabilized by the coordination force between the negative region of halogen atom and the gold atom, and the other contains a halogen bond formed between the σ-hole of the halogen atom and the negative electrostatic potential of Aun. Type I complexes are more stable than type II complexes, which is confirmed by geometric and energetic parameters. The result of electron density shift is consistent with the interaction energy. By means of AIM analysis, BCPs and RCPs were detected in type I and type II complexes respectively. The electron density of BCP in type I complexes is larger than that in type II complexes. The electronic energy density for these two types of complexes has different plus or minus, indicating different natures for the two noncovalent interactions.
References
Chalasinski G, Szczesniak MM (2000) Chem Rev 100:4227–4252
Rudkevich DM (2004) Angew Chem Int Ed 43:558–571
Saalfrank RW, Maid H, Scheurer A (2008) Angew Chem Int Ed 47:8794–8824
Metrangolo P, Resnati G (eds) (2007) Halogen bonding: fundamentals and applications, structure and bonding. Springer, Berlin
Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Model 13:291–296
Politzer P, Lane P, Concha MC, Ma YG, Murray JS (2007) J Mol Model 13:305–311
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748–7757
Politzer P, Riley KE, Bulat FA, Murray JS (2012) Comput Theor Chem 998:2–8
Politzer P, Murray JS (2013) Chem Phys Chem 17:278–294
Politzer P, Murray JS, Clark T (2013) Phys Chem Chem Phys 15:11178–11189
Politzer P, Murray JS (2013) Cryst Eng Commun 15:3145–3150
Metrangolo P, Resnati G (2001) Chem Eur J 7:511–2519
Corradi E, Meille SV, Messina MT, Metrangolo P, Resnati G (2000) Angew Chem Int Ed 39:1782–1786
Metrangolo P, Meyer F, Pilati T, Resnati G, Terraneo G (2008) Angew Chem Int Ed 47:6114–6127
Loc Nguyen H, Horton PN, Hursthouse MB, Legon AC, Bruce DW (2004) J Am Chem Soc 126:16–17
Legon AC (2010) Phys Chem Chem Phys 12:7736–7747
Metrangolo P, Neukirch H, Pilati T, Resnati G (2005) Acc Chem Res 38:386–395
Cavallo G, Metrangolo P, Pilati T, Resnati G, Sansotera M, Terraneo G (2010) Chem Soc Rev 39:3772–3783
Auffinger P, Hays FA, Westhof E, Ho PS (2004) Proc Natl Acad Sci U S A 101:16789–16794
Parisini E, Metrangolo P, Pilati T, Resnati G, Terraneo G (2011) Chem Soc Rev 40:2267–2278
Lu YX, Shi T, Wang Y, Yang HY, Yan XH, Luo XM, Jiang HL, Zhu WL (2009) J Med Chem 52:2854–2862
Lu YX, Wang Y, Zhu WL (2010) Phys Chem Chem Phys 12:4543–4551
Shirman T, Arad T, van der Boom ME (2010) Angew Chem Int Ed 46:926–929
Shirman T, Kaminker R, Freeman D, van der Boom ME (2011) ACS Nano 5:6553–6563
Blakey I, Merican Z, Rintoul L, Chuang YM, Jack KS, Micallef AS (2012) Phys Chem Chem Phys 14:3604–3611
Zhao Y, Truhlar DG (2006) J Chem Theory Comput 2:1009–1018
Zhao Y, Truhlar DG (2008) Acc Chem Res 41:157–167
Wang WZ, Zhang Y, Ji BM, Tian AM (2011) J Chem Phys 134:224303–224307
Zhang Y, Ma N, Wang WZ (2012) Chem Phys Lett 532:27–30
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Frisch MJ et al (2009) Gaussian 09 (revision B.01). Gaussian Inc, Pittsburgh
Bader RFW (1990) Atoms in molecules. A quantum theory. Oxford University Press, New York
Keith TA (2013) AIM All Version 13.05.06, aim.tkgristmill.com
Lu T, Chen FW (2012) J Comput Chem 33:580–592
Hammod RP, Cavaluzzi M, Haushalter RC, Zubieta JA (1999) Inorg Chem 38:1288–1292
Lu JY, Cabrera BR, Wang RJ, Li J (1999) Inorg Chem 38:4608–4611
Wang WZ, Wong NB, Zheng WX, Tian AM (2004) J Phys Chem A 108:1799–1805
Li QZ, Li R, Liu XF, Li WZ, Chen JB (2012) Chem Phys Chem 13:1205–1212
Acknowledgments
The author is grateful for the help of the high performance computing centre in Shandong University and reasonable advice of Prof. Feng in Shandong University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhao, Q. The X∙∙∙Au interactions in the CF3X (X = Cl, Br) ∙∙∙Aun (n = 2, 3, and 4) complexes. J Mol Model 20, 2133 (2014). https://doi.org/10.1007/s00894-014-2133-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-014-2133-8