
Abstract
We perform density functional theory studies to investigate structural and electronic properties of the (5,5) boron nitride nanotubes (BNNTs) with surfaces and ends functionalized by thiol (SH) and hydroxyl (OH) groups. The exchange-correlation energies are treated according to the functional of Hamprecht-Cohen-Tozer-Handy within the generalized gradient approximation (HCTH-GGA). We use the base function with double polarization DNP. To determine the (5,5) BNNT-SH and (5,5) BNNT-OH relaxed structures the minimum energy criterion is applied considering six different geometries depending upon the SH and OH functional groups orientation: (C1) The adsorbed functional group is oriented toward the N atom, (C2) the functional group is oriented toward the B atom, (C3) the functional group is at the central hexagon of the BNNT surface. The (C4) fourth and (C5) fifth configurations are formed by allowing bonds (of S or O) with B or N atoms at one end of the nanotube. (C6) The sixth geometry is obtained by placing the functional group at the center of one end of the BNNT. The (5,5) BNNT-SH system, in vacuum, suffers a semiconductor to metal transition while the (5,5) BNNT-OH system retains the semiconductor behavior. When structures are solvated in water these systems behave as semiconductors. The polarity increases as a consequence of the functional group-nanotube interactions no matter if they are in vacuum or in solvation situation, which indicates the possible solubility and dispersion. According to the work function the best option to construct a device is with the BNNT-OH system.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Since the theoretical prediction by Rubio et al. [1] and the synthesis by Chopra et al. [2] boron nitride nanotubes (BNNTs) have received a little attention of the scientific community both theoretical and experimentally. As a consequence BNNTs have not been explored in medical applications or new semiconductor devices. This is in part due to the low solubility and the difficulty to clean the surface of the system. However recent reports show that functionalized nanotube surfaces may be solvated in water [3]. BNNTs may have biological applications as an anticancer agent by reducing the toxicity [4]. When the nanotube surface is functionalized by polymers the nanotube may be employed as glucose biosensor [5] and visible light emitter device [6]. As it stands it is very important to investigate the functionalization of nanotube surface and ends (because of dangling bonds) by different functional groups to modify the electronic properties. Theoretical works [7–13] have reported variations of electronic and magnetic properties of BN nanotubes and nanosheets because of the functionalization by organic molecules or functional groups, which in turn indicate the way to explore nanotechnology. Therefore in this work we study the surface functionalization effects on the structural (bond length and nanotube diameter) and electronic properties (polarity, chemical potential, first ionization potential and work function) of single wall boron nitride nanotubes (SW-BNNT) having armchair structure with chirality (5,5). The functionalization of the surface and ends of the nanotubes is made by thiol (SH) and hydroxyl (OH) groups. Afterward we use molecular simulations to explore the solvation in water. This is done within the density functional theory which has been demonstrated to yield good results [14–19] for nitride compounds. One point to remark is that we also search for possible technological applications.
Simulation models and methods
First principles total energy calculations are performed to study the (5,5) boron nitride nanotubes functionalized by SH and OH (BNNT-X; X = SH and OH). Studies are done employing the density functional theory using the Hamprecht-Cohen-Tozer-Handy (HCTH) [20] functional within the generalized gradient approximation (GGA) with a double polarized base function DNP (that is we use a p orbital for hydrogen and a d for B, N, O, and S) [21] as implemented in the quantum chemistry DEMOL3 code [22]. For each functional group six different geometries are explored: (C1) in the first the adsorbed functional group is oriented toward the N atom, (C2) in the second the functional group is oriented toward the B atom, (C3) in the third the functional group is at the central hexagon of the BNNT surface. The (C4) fourth and (C5) fifth configurations are formed by allowing bonds between the functional group (S or O) and B or N atoms at one end of the nanotube. The (C6) sixth geometry is obtained by placing the functional group at the center of one end of the BNNT (Fig. 1). The nanotubes of chirality (5,5) have 1.32 nm of length and 0.77 nm of diameter are mono-hydrogenated at both ends (open nanotubes), they are formed by 120 atoms (50 N, 50 B and 20 H) and a functional group either thiol or hydroxyl. Neutral charge is present in the functionalized nanotubes and functional groups, and the multiplicity we have used is 1 for each analyzed configuration, that is, no spin has been taken into account.

We show the initial configurations of the interaction between BNNT and OH functional group. Similar geometries were used to study the interactions at the BNNT-SH system
Solvation effects are taken into account by using the polarized continuum model (conductor-like screening model) [23–25], in our studies we consider water as solvent with a dielectric constant of 78.4. The solvation energy difference are determined through the formulas Esolv1 = E(BNNTsolv)-E(BNNTvacuum) and Esolv2 = E[(BNNT-X; X = SH or OH)solv]-E[(BNNT-X;X = SH or OH)vacumm]. We have also obtained the ionization first potential using the formula I.P. = [E(BNNT-X;X = SH or OH)] + 1-[E(BNNT-X;X = SH or OH)]0 and the energy gap as the difference between HOMO and LUMO orbitals. The adsorption energy of the SH and OH functional groups is defined according to: Ead = E(BNNT + X;X = SH or OH)-E(BNNT)-E(X = SH or OH). The chemical potential is calculated as the arithmetic average (HOMO + LUMO)/2 provided that for the electron gas this is equal to the Fermi level and is considered as the center of the energy gap. The work function is determined as the energy difference of the potential energy at the vacuum (LUMO) and the Fermi level (chemical potential), which represents the minimum energy to remove an electron from the Fermi level to the vacuum level. The molecular electrostatic potential (MEP) is obtained as reported in the literature [26]. We have used the following cut radius for the atomic orbitals: 0.33 nm for the functional groups, 0.40 nm for the BNNT-OH system, 0.41 nm for BNNT and BNNT-SH, all these applied to the base function with a convergence tolerance of 1.0 × 10−6 Ha. To achieve the structural stability we have considered the non-complex vibration frequency [27] criterion.
Results and discussion
It is worthwhile noting that BN nanotubes with open ends and saturated dangling bonds by B or N may be studied using molecular simulation as already shown by the recent theoretical report of Hao et al. [28]. In this work magnetic properties studies have been performed on nanotubes with open ends in different chiralities. Other studies have reported good results for the energy gap of nanotubes with (n, n) chirality [29], with studies being important provided these nanotubes with open ends may be synthesized [30]. These nanotubes are appropriate to be functionalized by polymers which in turn allow the applications in biomedicine as they may exhibit high mechanical resistance [3, 4].
Analysis of the geometrical optimization process
We have determined the most stable atomic structures (see Table 1) of the interaction between the SH functional group and the nanotube of configuration C1 (Fig. 2a). Our studies consider the geometry with the functional group at the nanotube surface, which is different to that studied in the Zhao et al. [13] work which considers the molecule at the nanotube ends claiming that the interactions are stronger.

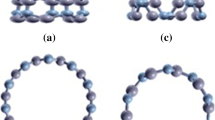
This figure displays the optimized atomic structures in (a) BNNT, (b) MEP for BNNT, (c) BNNT-SH, (d), MEPs for BNNT-SH, (e) BNNT-OH, (f) MEPs for BNNT-OH. In a), c) and e) the blue color represents N, pink is B, yellow is S, red is O and white is H. In b), d) and f) the blue color represents the positive charge and the yellow color the negative charge
It is important to remark that in metastable configurations of the BNNT-SH system a small diameter contraction takes place. In configuration C6, the SH functional group interacts with the end of the nanotube producing a reorientation of the thiol with the H atom directed toward the open end. According to Table 2 the metastable configuration C2 exhibits a bond between SH and the nanotube B atom, the system behaves very much like the optimal atomic structure. Changes in the atomic structure allow the thiol to remain a distance of 4.08 Å away from the nanotube. The distance is measured with respect to a nitrogen atom of a hexagon with the thiol oriented toward the center of the hexagon. The H-S bond length is 1.35 Å and the corresponding B-N bond length is 1.44 Å. These results are in agreement with those of the BN nanotube with no impurities. Note that the diameter of the nanotube in the BNNT-SH system is contracted by 0.01 nm. The adsorption energy of the thiol on the BNNT surface is −0.06 eV and provided that the van der Waals force is weak we obtain physisorption. The weak van der Waals interaction between the thiol and the BNNT is attributed to the low thiol polarity (0.74 D).
We now discuss results of the interaction between the functional group OH and BNNT. The configuration of the lowest minimum energy corresponds to geometry C2 (see Fig. 2e) where the functional group interacts with a B atom of the nanotube (see Table 1). The OH-B exhibits a bond length of 1.48 Å and the O-H displays a bond length of 0.96 Å. This OH group-BNNT interaction induces a protrusion on the bond which in turn modifies the hybridization. The nanotube diameter remains constant having a value 0.77 Å. We obtain chemisorption with energy of −7.90 eV.
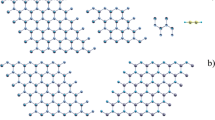
In configuration C3, a metastable structure, the nanotube is affected by the dissociation of the B-N bond as a consequence of the interaction with the OH group producing a vacancy and the formation of an octagon. It can be seen that the oxygen atom migrates into one nanotube octagon and the hydrogen into another octagon. The nanotube is also affected by a diameter reduction (see Fig. 3). This yields a possible way to generate defects in the nanotube structure such as vacancies in non-hexagonal atomic geometries. In contrast, in a similar configuration the interaction between SH and the BNNT the nanotube surface regenerates and expels the functional group. In experimental studies by Miramoto et al. [31] using ultrafast laser pulses (ULPs) HCl molecules have been incrusted into carbon and boron nitride nanotube surfaces in a perpendicular direction. Such studies motivate the possible application in nanochemical confinement or in the manipulation of molecules in nanotube structures.

This figure shows a section of configuration 3 (optimized), where we display changes in the atomic structure as a consequence of the interactions with the OH group at the surface of the BNNT. Blue spheres represent N; pink spheres represent B; red is O and white is H
Analysis of the electronic properties
The MEPs surfaces (Fig. 1b) show how the electron distribution is affected by the interaction of the functional group with the nanotube, this yields the electron charge concentration on the thiol (Fig. 2d) and on the hydroxyl (Fig. 2f). Note that the BNNT-SH system retains the BNNT characteristics provided the charge is also localized on the N as a consequence of the weak interaction with the thiol, meanwhile in the BNNT-OH structure the charge is mostly localized at the interaction site. It is important to remark that the electronic distribution of both systems is not localized at the ends of the nanotubes. One may conclude that it is possible to disperse low concentrations of these structures when placed in vacuum or in an organic solvent with negative net charge, in this way the possible technological applications may emerge.
On the other hand, the isolated BNNT exhibits a semiconductor behavior with an energy difference between the HOMO and LUMO orbitals of 4.78 eV. We shall mention that in our calculations we have used different approaches to deal with the exchange-correlation energies with the best results corresponding to the HTCH functional within the generalized gradient approximation (Table 2). The selection of this HTCH functional has been done after the BN nanosheets electronic and structural properties [32] studies considering several functional.
The interaction of the SH functional group with the BNNT induces a transition from a semiconductor to a metal, in contrast the OH group interacting with the BNNT makes only a small energy gap reduction (0.31 eV), the semiconductor characteristics remain (energy gap of 4.47 eV). The polarity of the BNNT (having covalent bonds) changes from the value of 0 D to a polarity of 3.71 D when SH is attached to the BNNT (resulting in ionic characteristics) meanwhile the OH group interacting with the BNNT induces a polarity of 3.11 D in the BNNT-OH system (with an ionic behavior). The charge is redistributed at the interaction site, that is, at the zone where the functional group interacts with the nanotube. This is clearly shown by the MEPs surfaces presented in Figs. 2d and 2f. The polarity increase is also manifested in the BN nanotubes with different chirality [9] as well as in BN nanosheets [11] when interacting with the natural biopolymer chitosan. The polarity is a suitable parameter to check the solubility of the BNNT-SH and BNNT-OH structures in a similar fashion as the solvation of BNNT--methoxy-poly (ethylene gycol)-1, 2-distearoyl-sn-glycero-3-phospoethanolamine-N in water [3]. Taking into account that the changes in the polarity are related to the solubility we performed calculations to simulate the solvation of both BNNT-SH and BNNT-OH systems in water. In the process we obtain an increase in the polarity as a result of the interactions of the functional groups with the nanotube. The polarity reaches the values of 11.80 D for the BNNT-SH system and of 7.51 D for BNNT-OH.
For biochemical applications it is important to learn about the solvation energy difference, therefore here we report the energy difference of the isolated BN nanotube Esolv = −227.38 kcal mol-1, of the BNNT-SH system Esolv = −22.37 kcal mol-1 and of the BNNT-OH system Esolv = −234.06 kcal mol-1. Note that the solvation energy difference corresponding to the BNNT-OH system becomes more negative by −6.68 kcal mol-1 as compared with the energy of the BNNT. This may be interpreted as a result of the conduction electron polarization. The increase in the solvation energy (becomes more positive) of the BNNT-SH system is produced by the weak interaction between the nanotube and the thiol. The solvation of BNNT-SH in water produces structural changes yielding a separation distance of 4.36 Å between the nanotube and the thiol (this distance is measured with respect to the N atom in the nanotube). At the same time the S-H bond length is increased to 1.35 Å, with the H atom oriented perpendicular to the BNNT surface. The BNNT-OH structure shows an increment in the O-B bond length taking the value of 1.48 Å but the O-H bond length remains unchanged.
The work function of every functionalized nanotube indicates that the charge transfer to the nanotube is low for the BNNT-OH (2.24 eV) as compared with that of the BNNT-SH (12.77 eV). This result means that the presence of adsorbates on the nanotube surface improves conditions for better field emission properties (FEPs) of the BNNT-OH system in agreement with reports in the literature concerning the interaction of the water molecule and oxygen molecule with BN nanotubes [13]. In these systems the potential barrier for the electron emission is reduced, consequently electrons are emitted from the BNNT surface and from both nanotube ends.
The ionization potential energy of the BN nanotube is 6.87 eV, of the BNNT-SH system is 6.49 eV and of the BNNT-OH system is 6.96 eV, which yield a variation of the order of 10−2 eV. A similar discussion can be given in terms of the chemical potential (Fermi energy level, see Table 2) provided that the corresponding BNNT-SH chemical potential is reduced (to −14.02 eV) as compared with that of isolated BNNT chemical potential (−3.65 eV). Meanwhile the chemical potential of the BNNT-OH system increases somewhat (−3.56 eV) with the chemical reactivity taking the value of 0.1 eV. We may conclude that the BNNT-OH system may be used technologically in the fabrication of optoelectronic devices. Results obtained in our work are similar to those of the solvation BNNTs in water (see Table 2). It is also important to note that solvation of the BNNT-SH system (having zero gap in vacuum, but having gap of 4.72 eV when solvated) induces no changes in the semiconductor behavior, in a similar fashion as the BNNT-OH system, however in this latter structure the solvation produces a small energy change of the order of 0.17 eV. Finally the electric conductivity is calculated according to the expression σ α exp (-Eg/kT), where k is the Boltzmann constant and T the temperature [36]. At a given temperature the BNNT-SH system displays a high conductivity provided the energy gap is small (in vacuum), on the other hand the BNNT-OH system with large energy gap exhibits kinetic stability in vacuum and solvated.
Conclusions
We have presented first principles total energy calculations to study the interactions between the SH and OH functional groups and the (5,5) boron nitride nanotubes (BNNTs). Results show that the functional groups and the nanotubes sustain chemical interactions. Moreover the functionalization is obtained on the nanotube surface instead of the ends. The BNNT-SH system exhibits changes with the most important being the transition from a semiconductor to a metal, while the BNNT-OH structure retains the semiconductor characteristics. The increase in the polarity of both systems indicates the possibility of solvation and dispersion as suggested also by the negative energy of solvation for the functionalized structures. Finally we find that the BNNT-OH system is the best option to be used in the possible fabrication of devices such as the display by means of the FEPs. This proposal is supported by the low adsorption energy and work function.
References
Rubio A, Corkill JL, Cohen M (1994) Phys Rev B 49:5081–5084
Chopra NG, Luyken RJ, Cherrey K, Crespi VH, Cohen ML, Louie SG, Zetl A (1995) Science 269:996–967
Huei Lee C, Zhang D, Khin Yap Y (2012) J Phys Chem C 116:1798–1804
Raffa V, Riggio C, Smith MW, Jordan KC, Cao W, Cuschieri A (2012) Technol Cancer Res Treat 11:459–527
Wu J, Yin L (2011) ACS Appl Mater Interfaces 3:4354–4362
Gao Z, Zhi C, Bando Y, Golberg D, Serizawa T (2011) ACS Appl Mater Interfaces 3:627–632
Zhi CY, Bando Y, Tang CC, Honda S, Sato K, Kuwahara H, Golberg D (2005) Angew Chem Int Ed 44:7929–7932
Xie SY, Wang W, Fernando KAS, Wang X, Lin Y, Sun YP (2005) Chem Commun 29:3670–3672
Rodríguez Juárez A, Chigo Anota E, Hernández Cocoletzi H, Flores Riveros A (2012) Appl Surf Sci. doi:10.1016/j.apsusc.2012.12.075
Rodríguez Juárez A, Chigo Anota E, Hernández Cocoletzi H (2013) Structural and electronic properties of boron nitride nanotubes-chitosan: effect of point defects. J Mol Graph Model. (in press)
Chigo Anota E, Hernández Rodríguez L D, Rodríguez Juárez A (2013) Appl Surf Sci. (in press)
Chigo Anota E, Rodríguez Juárez A, Castro M, Hernández Cocoletzi H (2013) J Mol Model 19(1):321–328
Zhao J X, Ding Y H (2009) Nanotechnol 20: 085704(1)-(6)
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi H (2011) J Nanosci Nanotechnol 11(6):5515–5518
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi H (2010) Phys Stat Solidi C 7(7–8):2252–2254
Chigo Anota E, Salazar Villanueva M, Hernández Cocoletzi H (2010) Phys Stat Solidi C 7(10):2559–2561
Chigo Anota E, Hernández Cocoletzi H, Rubio Rosas E (2011) Eur Phys J D 63:271–273
Chigo Anota E, Ramírez Gutierrez RE, Escobedo Morales A, Hernández Cocoletzi G (2012) J Mol Model 18(5):2175–2184
Galícia Hernández JM, Hernández Cocoletzi G, Chigo Anota E (2012) J Mol Model 18(1):137–144
Boese AD, Handy NC (2001) J Chem Phys 114:5497–5503
Delley B (1990) J Chem Phys 92:508–517
Delley B (2000) J Chem Phys 113:7756–7765
Klamt A, Schüürmann G (1993) J Chem Soc Perkin Trans 2:799–805
Delley B (2006) Mol Simul 32:117–123
Tomasi J, Persico M (1994) Chem Rev 94:2027–2094
Hernández Rosas JJ, Ramírez Gutiérrez RE, Escobedo Morales A, Chigo Anota E (2011) J Mol Model 17(5):1133–1139
Foresman JB, Frisch AE (1996) Exploring chemistry with electronic structure methods. Gaussian Inc, Pittsburgh, p 70
Hao S, Zhou G, Duan W, Wu J, Gu BL (2006) J Am Chem Soc 128:8453–8458
Xiang H J, Yang J, Hou J G, Zhu Q (2003) Phys Rev B 68: 035427(1)-(5)
Golberg D, Bando Y (2001) Appl Phys Lett 79:415–41
Miyamoto Y, Zhang H, Rubio A (2010) Phys Rev Lett 105: 248301(1)-(4)
Chigo Anota E, Santos Castillo JR (2013) Structural and electronic properties of the boron nitride nanosheets functionalized with thiol and hydroxyl groups. J Comp Theor Nanosci. (in press)
Kang HS (2006) J Phys Chem B 110:4621–4628
Blase X, Rubio A, Louie SG, Cohen ML (1994) Europhys Lett 28:335–340
Arenal R, Stephan O, Kociak M, Taverna D, Loiseau A, Colliex, C (2005) Phys Rev Lett 95:127601(1)-(4)
Li S (2006) Semiconductor physical electronics 2nd edn. Springer, Berlin
Acknowledgments
This work was partially supported by projects: VIEP-BUAP (CHAE-ING13-G), Cuerpo Académico Ingeniería en Materiales (BUAP-CA-177), Cuerpo Académico Física Computacional de la Materia Condensada (BUAP-CA-194) and VIEP-BUAP-EXC11-G.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chigo Anota, E., Cocoletzi, G.H. First-principles simulations of the chemical functionalization of (5,5) boron nitride nanotubes. J Mol Model 19, 2335–2341 (2013). https://doi.org/10.1007/s00894-013-1782-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1782-3