
Abstract
To compare the OH-initiated reaction mechanisms of five chlorophenoxy herbicides, density functional theory (DFT) calculations of reactions in which ·OH attacks one of three active positions on each herbicide were carried out at the MPWB1K/6-311 + G(3df,2p)//MPWB1K/6-31 + G(d,p) level. For each herbicide, the calculation results show that ·OH addition to the C1 atom, which is the nexus between the benzene ring and the side group, possesses the lowest energy barrier among the three kinds of reactions, indicating that ·OH addition–substitution of the side chain is the most energetically and kinetically favorable reaction mechanism. Comparisons among the herbicides show that the mechanisms are affected by the steric hindrance and the electronegativities of the –CH3 and –Cl groups. When comparing the addition of ·OH to the C1 site among the five herbicides, the activation energy for the reaction of ·OH with DCPP reaction is the lowest (3.61 kcal mol−1), while that for the ·OH and 4-CPA reaction was the highest (5.91 kcal mol−1). ·OH addition to the C4 site presents the highest energy barriers among the three kinds of reactions, indicating that the para Cl is difficult to break down. When comparing the H-atom abstraction reactions of the five herbicides, the H atoms in the –CH2– group of 2,4-D are the easiest for ·OH to abstract, whereas those of DCPP and MCPP are more difficult to abstract, due to the steric hindrance of the –CH3 group. Additionally, the results obtained from the PCM calculations reveal that most of the reactions occur more easily in water than in gas, though the mechanisms involved are the same as those discussed above.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Herbicides—a large class of pesticides that contribute greatly to water contamination—have been used extensively in agriculture since World War II [1–3]. Among the various kinds of herbicides available, 4-chlorophenoxyacetic acid (4-CPA), 2,4-dichlorophenoxyacetic acid (2,4-D), 4-chloro-2-methylphenoxyacetic acid (MCPA), 2-(2,4-dichlorophenoxy)propanoic acid (DCPP), and 2-(4-chloro-2-methylphenoxy)propanoic acid (MCPP)—all of which are chlorinated phenoxy acids—are used on a large scale to control the growth of broadleaf weeds in fields growing cereals, grasses, fruits, tobacco, and cotton [1, 4–10].
Due to their intensive use in agriculture, most herbicides penetrate into the soil or enter surface water via runoff from agricultural lands [1, 11, 12]. As a result, relatively high concentrations of these herbicides are detected in groundwater and surface water; indeed, they are regarded as the most frequently detected organic substances in such waters [13–16]. In addition, these herbicides are moderately toxic (class II or III) to humans and animals (according to the World Health Organization [4, 9, 17, 18]), and take a long time to degrade in the environment [19–21].
For these reasons, there is considerable interest in the degradation of chlorophenoxyl herbicides using advanced oxidation processes (AOPs), such as photochemical [12, 22–25], photocatalytic [14, 26–33], and electrochemical [9, 17, 34–37] methods, based on the generation of a powerful oxidant agent—the hydroxyl radical (·OH). Because these techniques are very efficient at degrading chlorophenoxyl herbicides, many researchers are trying to develop new catalysts and to improve electrodes in order to increase their oxidizing power. Moreover, most research in this field [29, 31–33, 36] has focused on laboratory experiments, and some simple degradation mechanisms have been proposed [9, 14, 24, 26], but these are not sufficient to gain a detailed understanding of the degradation mechanism. Additionally, little information on the differences among the degradation mechanisms of the five herbicides (with similar structures) mentioned above has been reported. Therefore, a theoretical study of the OH-induced degradation mechanisms for these five herbicides is necessary.
According to previous experiments [14, 24, 38, 39], the most active sites in these herbicides are the ipso and para C sites and the H atom in the –CHR– group of the side chain. Furthermore, the results of our theoretical calculations show that the reactions that occur at the above sites in MCPA all have low energy barriers, and are thus energetically favorable [40]. Therefore, in the work described in the present paper, DFT calculations were performed in order to compare the different reaction mechanisms of the five herbicides corresponding to ·OH attacks at the three active sites. Studying these reaction pathways in detail allowed us to determine which of the active sites corresponds to the most kinetically favorable mechanism for each herbicide, and to suggest factors that probably lead to mechanistic differences for the same type of reaction among the five herbicides.
Computational methods
The geometries of all stationary points (including reactants, transition states, intermediates, and products) involved in the addition and abstraction reactions were optimized at the MPWB1K/6-31 + G(d,p) level [41] in the framework of DFT using the Gaussian 03 package [42]. MPWB1K/6-31 + G(d,p) is considered to be an excellent method to use to predict thermochemical kinetics, hydrogen bonding, transition state geometries, and vibrational frequencies of chlorinated organic compounds [43–45]. The nature (local minimum or first-order saddle point) of each stationary point was identified by performing harmonic vibrational frequency calculations at the same level at which the zero-point energies (ZPEs) were also derived. All transition states were characterized by one negative eigenvalue. It was also verified that all transitions connect the corresponding reactants with the products by performing intrinsic reaction coordinate (IRC) [46, 47] calculations. Moreover, single-point energy calculations were carried out at the MPWB1K/6-311 + G(3df,2p) level to obtain more accurate energy parameters based on the structures optimized at the MPWB1K/6-31 + G(d,p) level. In the present study, to estimate the solvent effects for all pathways, the polarizable continuum model (PCM) with water as solvent was employed, based on the optimized gas-phase structures.
Results and discussion
The mechanisms of ·OH attack at the three selected active sites for each herbicide (addition–substitution of the side chain, addition–substitution of the para Cl, and H-atom abstraction) were simulated through DFT calculations.
Molecular structures of the five herbicides
The optimized molecular structures of and atom numbering schemes used for 4-CPA (I), 2,4-D (II), MCPA (III), DCPP (IV), and MCPP (V) are shown in Fig. 1. As illustrated in Fig. 1, the molecular structures of 4-CPA, 2,4-D, and MCPA are planar, except for the aliphatic atoms. Due to the existence of the –CH3 group attached to C8, the main bodies of DCPP and MCPP are not planar; the dihedral angles ϕ 1 (C1–O1–C8–C9) and ϕ 2 (C1–O1–C8–C9) are 20.2° and 24.5°, respectively. However, the basic structures of the five compounds are similar, with a benzene ring and a –COOH group. Upon comparing the other molecules with 4-CPA, we can see that the ortho H atom is substituted for (i) a Cl atom in 2,4-D and (ii) a –CH3 group in MCPA. A –CH3 group is substituted for (i) one of the H atoms attached to C7 in 2,4-D to give DCPP, and (ii) the C8 atom in MCP to give MCPP.

The optimized molecular structures of and atomic numbering schemes used for 4-CPA, 2,4-D, MCPA, DCPP, and MCPP
Addition–substitution mechanisms of the side-chain group
Five channels (denoted I-R1 to V-R1) corresponding to ·OH addition to C1 in the five herbicides were identified and are shown in Scheme 1, and all of the relative energies for these channels (including reaction enthalpies and heats) are displayed in Table 1. The optimized geometries, including the intermediates (IM) and transition states (TS) involved in the reactions, are displayed in Fig. 2a. Profiles of the potential energy surface corrected for zero-point energy (ZPE) are presented in Fig. 3a. The main products are shown in Fig. S1 (in the “Electronic supplementary material,” ESM). It is worth noting that the structures obtained for MCPA were illustrated in a previous paper [40].

Possible pathways for ·OH addition to C1 in the five herbicides




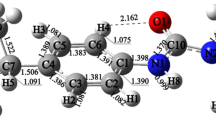
Optimized geometries of the intermediates (IM) and transition states (TS) for the reactions of the five herbicides with ·OH calculated at the MPWB1K/6-31 + g(d,p) level. a Geometries for ·OH addition to C1; b geometries for ·OH addition to C4; c geometries for H-atom abstraction. The gray, white, red, and green balls denote carbon, hydrogen, oxygen, and chlorine atoms, respectively, and bond lengths are shown in angstroms

Calculated potential energy surface profiles for all reaction pathways: a ·OH addition to C1; b ·OH addition to C4; c H-atom abstraction
As shown in Scheme 1, the reaction patterns for the five channels are similar, so we will now discuss the mechanism of ·OH addition to C1 using channel I-R1 as an example. From Fig. 2a and Fig. 3a, it can be seen that channel I-R1 proceeds with an energy barrier of 5.91 kcal mol−1 via transition state I-TS1, where ·OH is attacking 4-CPA from a direction almost perpendicular to the molecule (the angle O–C1–O1 is 95.9°). An adduct, I-IM1, is then found on the potential energy surface. Compared with those in I-TS1, the distance C1…O is shorter and the bond C1–O1 is longer, indicating that the bond C1–O is being formed and the bond C1–O1 is being broken. Finally, the unimolecular decomposition of I-IM1 leads to 4-chlorophenol (denoted P1) and · OCH2COOH. The overall reaction is exothermic by 13.07 kcal mol−1. The main product is 4-chlorophenol, in accordance with experimental observations [9, 26, 38]. It is interesting to note that 4-chlorophenol is an important precursor for the formation of PCDD/Fs and, according to information in [48], the reaction of 4-chlorophenol with ·OH has a lower activation energy, indicating that 4-chlorophenol is easier to degrade than 4-CPA is. Moreover, the detailed mechanism of the reaction of 4-chlorophenol with ·OH has been investigated by many workers [48–50].
Reactions II-R1, III-R1, IV-R1, and V-R1 proceed in a similar way to the reaction I-R1. The dominant product for reactions II-R1 and IV-R1 is 2,4-dichlorophenol (denoted P2), which is in good agreement with experimental results [12, 27, 46–52]. For reactions III-R1 and V-R1, the main product is 4-chloro-2-methylphenol (denoted P3), which again matches well with experimental observations [6, 14, 24, 25, 39, 51].
As illustrated in Fig. 2a, no pre-complex is formed in each channel. The structures of all five transition states were found to be similar. The C1…O distances in the transition states (∼2.0 Å) are longer than the usual C–O bond distance (∼1.4 Å) [52], indicating that a transition state is formed early on in each reaction, in accord with Hammond’s postulate [52]. The basic structures of all of the adducts are similar to those of the corresponding transition states. However, the C1…O distances become much shorter, decreasing from about 2.0 Å in the transition states to about 1.4 Å, and all of the C–C1–O angles become close to 110°, as expected for sp 3-hybridized carbon [52].
Though they do show some similarities, the five reactions differ in the potential energies and structures associated with them. As Table 1 summarizes, pathway IV-R1 has the lowest activation energy (3.61 kcal mol−1, followed by V-R1 (4.06 kcal mol−1), III-R1 (4.83 kcal mol−1), II-R1 (5.38 kcal mol−1), and I-R1 (5.91 kcal mol−1). These energy differences mainly result from the different electronegativities of the substituent groups. The effects of the –Cl and –CH3 groups on the mechanisms for the five reactions were compared on the basis of the structures and energy parameters involved. Comparing channel II-R1 with channel I-R1, the C1–O bond that is being formed in II-IM1 is 0.01 Å shorter than that in I-IM1, and the angle O–C1–O1 in II-IM1 is 0.7° smaller than that in I-IM1. This result indicates that II-IM1 is slightly more like the product than I-IM1 is. As shown in Table 1, the energy barrier of 5.38 kcal mol−1 for channel II-R1 is lower than that (5.91 kcal mol−1) for channel I-R1, indicating that the C1 site of 2,4-D is easier for ·OH to attack than that of 4-CPA. This result is contrary to the electron-withdrawing nature of an ortho Cl substituent. Therefore, the reason for this behavior must be clarified. Comparing reactions III-R1 and II-R1, the C1–O bonds in III-TS1 and III-IM1 are both slightly longer than those in II-TS1 and II-IM1, respectively. This possibly results from the greater steric hindrance of the –CH3 group than the –Cl group. From Table 1, the barrier height for the reaction III-R1 is 0.55 kcal mol−1 lower than that for the reaction II-R1, indicating that the reaction III-R1 is more energetically favorable. This is probably because MCPA has an ortho –CH3 group with electron-donating characteristics, which can activate the aromatic ring and thus facilitate the reaction, while 2,4-D has an electron-withdrawing substituent (ortho Cl), which acts to deactivate the benzene ring. The electron-donating effect of the –CH3 group on the reaction must also be greater than that of the steric hindrance of the –CH3 group. Due to the presence of the electron-donating –CH3 group, the energy barrier for the channel IV-R1 is lower than that for the channel II-R1. Similarly, due to the presence of the electron-donating –CH3 group, the reaction between MCPP and ·OH occurs more easily than the reaction between MCPA and ·OH. The potential energy of the pathway IV-R1 is the smallest among the five reactions. This result indicates that the addition of ·OH to the C1 atom of DCPP is the most kinetically and energetically favorable of the five pathways. However, according to the activating/deactivating rule described above, pathway V-R1 should be most energetically favorable, although the activation energy is slightly higher than that of the pathway IV-R1. It is plausible that the combined steric hindrance of the two –CH3 groups in MCPP makes the reaction more unlikely to occur.
Furthermore, we can see from Table 1 that, for all five herbicides, the activation energies for ·OH addition to C1 are the lowest among all three types of reactions. This result indicates that C1 is the most active position in each herbicide. This fact is consistent with experimental results, in which the primary product results from ·OH substitution of the side chain [24, 53].
Addition–substitution mechanisms of the para Cl atom for the five herbicides
Five pathways (denoted I-R2 to V-R2) corresponding to ·OH addition to C4 in the five herbicides were identified and are presented in Scheme 2, and all of the relative energies (including reaction enthalpies and heats) for these are displayed in Table 1. The dominant products are shown in Fig. S1 of the ESM. The optimized geometries of the intermediates and the transition states involved in the reactions are shown in Fig. 2b, and the profiles of the potential energy surface corrected for zero-point energy (ZPE) are displayed in Fig. 3b.

Possible pathways for ·OH addition to C4 in the five herbicides and P3
As shown in Scheme 2, the five pathways I-R2 to V-R2 react in a similar way. Taking pathway I-R2 as example, we now discuss the reaction mechanism of ·OH addition to the C4 site. As illustrated in Scheme 2 and Fig. 2b, ·OH adds to the C4 atom via the transition state I-TS2 in the pathway I-R2. In the structure of I-TS2, the bond C4–Cl and the benzene ring are no longer in the same plane but are at an angle of 16.41° to each other, and the angle O–C4–Cl is 95.0°: almost a right angle. An intermediate, I-IM2, is then formed. A significant change occurs in the C4…O distance, which shortens from 1.983 Å in I-TS2 to 1.397 Å in I-IM2. A change is also seen in the angle between the C4–Cl bond and the benzene ring plane, which is 2.8 times larger than that in I-TS2. The shortened C4–O bond and the elongated C4–Cl bond in I-IM2 indicate that the C4–O bond will be formed and the C4–Cl bond will be broken. We can also see that all of the C4 atoms convert from sp 2 hybridization to sp 3 hybridization. Finally, the unimolecular decomposition of I-IM2 yields 4-hydroxyphenoxy acetic acid (denoted P4) and · Cl. The energy barrier for this pathway is 9.38 kcal mol−1, and the overall reaction is exothermic by 12.41 kcal mol−1. Pathways II-R2, III-R2, IV-R2, and V-R2 evolve similarly to pathway I-R2. The main products of reactions II-R2, III-R2, IV-R2, and V-R2 are 2-chloro-4-hydroxyphenoxy acetic acid (denoted P5), 4-hydroxy-2-methylphenoxyacetic acid (denoted P6), 2-chloro-4-hydroxyphenoxy propanoic acid (denoted P7), and 4-hydroxy-2-methylphenoxy propanoic acid (denoted P8), respectively.
From Fig. 2b, it is clear that no pre-complexes occur in the five pathways. All transition state structures are quite similar. The C4…O distances range from 1.978 Å to 1.988 Å, the C4…Cl distances range from 1.726 Å to 1.728 Å, and the O–C4–Cl angles are about 95°. The basic structures of all five intermediates are quite analogous. The ·OH is inserting itself onto the C4 atom, as indicated by the C4–O bond distance shortening, leading to the usual C–O bond. At the same time, the Cl atom is leaving the C4 atom. This fact is shown by the lengthening of the C4–C1 bond.
As illustrated in Table 1, the energy barriers increase successively from I-R2 to V-R2. This fact correlates well with the complexity of the steric configurations of the five herbicides. However, there is only 1.55 kcal mol−1 between the maximum observed for the reaction of MCPP and ·OH (10.93 kcal mol−1) and the minimum observed for the reaction of 4-CPA and ·OH (9.38 kcal mol−1). The effects of the –Cl and –CH3 groups on the mechanisms for the five reactions are small because these groups are in the ortho/para position. We can therefore conclude that the steric configuration is more complicated and that this reaction is less likely to occur. Additionally, we can see from Table 1 that the energy barriers for the addition of ·OH to C4 are generally the highest, indicating that the para Cl atom is difficult to substitute. This is in agreement with the fact that chlorinated organic compounds are difficult to degrade in natural environmental media [54–58].
According to previous research [4, 24], the substitution of the para Cl occurs after the substitution of the side chain. Therefore, if we take ·OH substitution of the para Cl from 4-chloro-2-methylphenol (P3) as an example (denoted Ra, see Scheme 2), the potential energy surface for this reaction is calculated to be comparable with that for the direct substitution of the para Cl from MCPA. The optimized structures, including the transition states and intermediates, are shown in Fig. S4 of the ESM. The calculated energy barrier is 8.33 kcal mol−1 for the reaction Ra. This is lower than that of the reaction III-R2, 10.29 kcal mol−1. This indicates that the Cl atom in 4-chloro-2-methylphenol is a little easier to substitute than that in MCPA. Therefore, the para Cl can be replaced by ·OH after the side chain.
H-atom abstraction mechanisms for five herbicides
Scheme 3 shows five channels (denoted I-R3 to V-R3) corresponding to the abstraction by ·OH of hydrogen atoms attached to the C7 atom of 4-CPA and 2,4-CD or hydrogen atoms attached to the C8 atom of MCPA, DCPP, and MCPP. All of the optimized structures involved in the five reactions are shown in Fig. 2c. The calculated potential energy surface profiles for H-atom abstraction are displayed in Fig. 3c. The main products P9–P13 are presented in Fig. S1 of the ESM.

Possible pathways for H-atom abstraction in the five herbicides
It can be seen from Scheme 3 that the five channels denoted I-R3 to V-R3 proceed via the same pattern. We will take channel I-R3 as an example to explain the H-atom abstraction mechanism. From Fig. 2c and Fig. 3c, it can be noted that a van der Waals pre-complex, I-IM3, occurs on the potential energy surface for channel I-R3. The energy of I-IM3 is 4.26 kcal mol−1 lower than that of the original reactants, meaning that the pre-complex is more stable. A transition state, I-TS3, is then formed upon surmounting an energy barrier of 9.06 kcal mol−1. In the structure of I-TS3, the C7–H5 bond is elongated by 7.24% and the O–H5 bond is reduced by 39.17% compared with those in I-IM3, respectively. An intermediate, I-IM4, is then formed, where the H2O generated previously is leaving the main body, and a weak H-bond is formed between H5 and the O atom of the –COOH group. Our IRC calculations also show an obvious H-atom transfer from C7 to the O atom. Finally, the unimolecular decomposition of I-IM4 produces the radical ·C6H4ClOCHCOOH (P9) and H2O. The overall reaction is strongly exothermic by 29.78 kcal mol−1. In a similar way, each of the channels II-R3, III-R3, IV-R3, and V-R3 evolves via a pre-complex, a transition state, an adduct and products, which are all found on the corresponding potential energy surface. The primary products for channels II-R3, III-R3, IV-R3, and V-R3 are ·C6H3Cl2OCHCOOH (P10), ·C6H4ClCH3OCHCOOH (P11), ·C6H3Cl2OCHCH3COOH (P12), and ·C6H4ClCH3OCCH3COOH (P13), respectively. Some of the products have been found as reactive intermediates in some studies [14, 59].
As illustrated in Fig. 2c, a pre-complex is first found in each pathway. The structure of IV-IM3 is different from those of the other four pre-complexes. Possibly, the combination of the –CH3 and the ortho –Cl in DCPP leads to the divergence of the –COOH group from the main plane, and finally a hydrogen bond between the H atom of ·OH and the O2 atom is formed in IV-IM3. IV-IM3 occurs since its energy is a little more stable than the other pre-complexes. Transition states I-TS3 to V-TS3 show obvious H-atom transfer from C7 to O for 4-CPA and 2,4-D, and from C8 to O for MCPA, DCPP, and MCPP. In the structures of I-IM4, II-IM4, III-IM4, IV-IM4, and V-IM4, the resulting H2O molecule leaves the main body, as demonstrated by the obviously stretched C–H bonds (C7–H5 for I-IM4, C7–H4 for II-IM4, and C8–H7 for III-IM4, for IV-IM4, and for V-IM4).
From Table 1, it can be seen that pathway II-R3 has the lowest energy barrier of 8.98 kcal mol−1, followed by I-R3, III-R3, V-R3, and IV-R3. This result suggests that the H atom in the –CH2- group of 2,4-D is the easiest to abstract. Additionally, the energy barriers for reactions IV-R3 and V-R3 are higher than those for reactions I-R3, II- R3, and III- R3. This result indicates that H atoms in the –CH2– group are easier to abstract than the H atom in the –CHCH3– group. This is probably due to the fact that the steric hindrance of the –CH3 group in the –CHCH3– group is greater than that of the H atom. The five abstraction reactions are all strongly exothermic, by around 30 kcal mol−1.
Water solvent effects on the degradation mechanism of each reaction
To demonstrate possible solvent dependences for all of the reactions, the results obtained from the PCM calculations are also shown in Table 1. The optimized geometries of the transition states gained from PCM calculations performed with water as solvent are presented in Fig. 4. The intermediates and the profiles of the potential energy surface corrected for the zero-point energy (ZPE) are depicted in Figs. S2 and S3 of the ESM, respectively.


Optimized geometries of the transition states in the reactions of the five herbicides with ·OH, calculated at the MPWB1K/6-31 + g(d,p) level with PCM: a for ·OH addition to the C1 atom; b for ·OH addition to the C4 atom; c for H-atom abstraction. The gray, white, red, and green balls denote carbon, hydrogen, oxygen, and chlorine atoms, respectively, and the bond lengths are shown in angstroms
As illustrated in Table 1, the calculated energy barriers to the substitution of the side chain in the five herbicides in water as solvent are slightly higher than those calculated in the gas phase. Figure 4a shows that the C1…O distances in all of the transition states are longer than those in the corresponding optimized structures in the gas phase. This is probably the reason that the addition of ·OH to the C1 site is a little more difficult to achieve in the water phase than in the gas phase. Moreover, the C1–O1 bond length in the transition state is similar whether the state is optimized in gas or in water (Figs. 2a and 4a). Figure S2 of the ESM indicates that the C1–O bond lengths in all adducts are slightly shorter than those in the corresponding geometries in the gas phase. These results match well with the results that the calculated energies of the adducts in the water phase are all smaller than those of the corresponding adducts in the gas phase, which indicates that the adducts are more stable in water than in gas.
From Table 1, it is apparent that the calculated barrier heights to the substitution of the para Cl atom in the five herbicides in water are a little smaller than those calculated in the gas phase. This means that the reactions occur more easily in water solvent. From Fig. 4b, it is clear that the C4–Cl bonds in the transition states are all longer than those in the gas phase, indicating that the para Cl atom is easier to substitute in the water phase than in the gas phase. It is interesting to note that all of the C4–Cl bonds are broken in adducts I′-IM2 to V′-IM2 in the water phase, whereas they are still intact (albeit elongated) in I-IM2 to V-IM2 in the gas phase (see Fig. S2 of the ESM).
Additionally, it can be noted from Table 1 that the activation energies for the H-atom abstraction reactions in water are slightly smaller than those in the gas phase. This fact indicates that the H atom is a little easier for ·OH to abstract in water than in gas. As shown in Fig. S3(c) in the ESM, a pre-complex is also formed initially during each reaction in the water phase. It also can be seen from Fig. 2c and Fig. 4c that in the transition states, the hydrogen bond between the O atom of ·OH and the H atom being abstracted is longer in the water phase than in the gas phase. This result is consistent with the fact that the calculated energies of the transition states in the water phase are higher than those of the transition states in the gas phase.
Conclusions
Three kinds of OH-initiated mechanisms for the degradation of five herbicides were compared on the basis of results obtained using the MPWB1K method. Comparisons reveal that the reaction mechanisms for the herbicides are affected by the different molecular structures induced by the –CH3 and –Cl groups. For each herbicide, the addition of ·OH to the C1 atom is the most kinetically favorable of the three kinds of reactions. For addition–substitution of the side-chain group, the energy barrier for the IV-R1 reaction is the lowest among the five reactions, probably due to a combination of the steric hindrance from and the electron-donating nature of the –CH3 group. The barrier heights associated with addition–substitution of the para Cl atom are generally the highest among the three kinds of reactions, indicating that the para Cl atom is difficult to substitute. Comparison of the direct addition–substitution of the para Cl atom in the original reactants with the addition–substitution in the primary products indicates that the substitution of the para Cl is easier to achieve after substituting the side chain. For the abstraction reactions, steric hindrance from the –CH3 group probably blocks the abstraction of the H atom by ·OH, meaning that the H atom in the –CH2– group is easier to abstract than that in the –CHCH3– group.
The PCM calculations reveal that the reactions occur more easily in water than in gas, except for the addition of ·OH to the C1 atom. A possible reason for this is that the longer C1…O distances in all the transition states in water phase than those in the corresponding optimized structures in gas phase makes it more difficult for ·OH to insert onto the C1 atom.
References
Gimeno O, Plucinski P, Kolaczkowski S, Rivas F, Alvarez P (2003) Removal of the herbicide MCPA by commercial activated carbons: equilibrium, kinetics, and reversibility. Ind Eng Chem Res 42:1076–1086
Thorstensen CW, Christiansen A (2001) Determination of bentazone, dichlorprop, and MCPA in different soils by sodium hydroxide extraction in combination with solid-phase preconcentration. J Agric Food Chem 49:4199–4202
Cabras P, Angioni A (2000) Pesticide residues in grapes, wine, and their processing products. J Agric Food Chem 48:967–973
CrespÍn MA, Gallego M, Valcárcel M (2001) Study of the degradation of the herbicides 2,4-D and MCPA at different depths in contaminated agricultural soil. Environ Sci Technol 35:4265–4270
Romero E, Dios G, Mingorance MD, Matallo MB, Peña A, Sánchez-Rasero F (1998) Photodegradation of mecoprop and dichlorprop on dry, moist and amended soil surfaces exposed to sunlight. Chemosphere 37:577–589
Flox C, Garrido JA, Rodríguez RM, Cabot P-L, Centellas F, Arias C, Brillas E (2007) Mineralization of herbicide mecoprop by photoelectro-Fenton with UVA and solar light. Catal Today 129:29–36
García-Campaña AM, Aaron J-J, Bosque-Sendra JM (2001) Micellar-enhanced photochemically induced fluorescence detection of chlorophenoxyacid herbicides. Flow injection analysis of mecoprop and 2,4-dichlorophenoxyacetic acid. Talanta 55:531–539
Kurt-Karakus PB, Bidleman TF, Muir DCG, Struger J, Sverko E, Cagampan SJ, Small JM, Jantunen LM (2010) Comparison of concentrations and stereoisomer ratios of mecoprop, dichlorprop and metolachlor in Ontario streams, 2006–2007 vs. 2003–2004. Environ Pollut 158:1842–1849
Boye B, Dieng MM, Brillas E (2002) Degradation of herbicide 4-chlorophenoxyacetic acid by advanced electrochemical oxidation methods. Environ Sci Technol 36:3030–3035
Bojanowska-Czajka A, Drzewicz P, Zimek Z, Nichipor H, Nalecz-Jawecki G, Sawicki J, Kozyra C, Trojanowicz M (2007) Radiolytic degradation of pesticide 4-chloro-2-methylphenoxyacetic acid (MCPA)—experimental data and kinetic modelling. Radiat Phys Chem 76:1806–1814
Vizcaíno P, Pistocchi A (2010) A GIS model-based assessment of the environmental distribution of [gamma]-hexachlorocyclohexane in European soils and waters. Environ Pollut 158:3017–3027
Kundu S, Pal A, Dikshit AK (2005) UV induced degradation of herbicide 2,4-D: kinetics, mechanism and effect of various conditions on the degradation. Sep Purif Technol 44:121–129
Henriksen T, Svensmark B, Lindhardt B, Juhler RK (2001) Analysis of acidic pesticides using in situ derivatization with alkylchloroformate and solid-phase microextraction (SPME) for GC-MS. Chemosphere 44:1531–1539
Topalov A, Abramovic B, Molnár-Gábor D, Csanádi J, Arcson O (2001) Photocatalytic oxidation of the herbicide (4-chloro-2-methylphenoxy)acetic acid (MCPA) over TiO2. J Photochem Photobiol A 140:249–253
Donald DB, Cessna AJ (2007) Pesticides in surface drinking-water supplies of the Northern Great Plains. Environ Health Perspect 115:1183
Rügge K, Juhler RK, Broholm MM, Bjerg PL (2002) Degradation of the (R)- and (S)-enantiomers of the herbicides MCPP and dichlorprop in a continuous field-injection experiment. Water Res 36:4160–4164
Brillas E, Boye B, Sirés I, Garrido JA, Rodríguez RM, Arias C, Cabot P-L, Comninellis C (2004) Electrochemical destruction of chlorophenoxy herbicides by anodic oxidation and electro-Fenton using a boron-doped diamond electrode. Electrochim Acta 49:4487–4496
Hoar SK, Blair A, Holmes FF, Boysen CD, Robel RJ, Hoover R, Fraumeni JF (1986) Agricultural herbicide use and risk of lymphoma and soft-tissue sarcoma. JAMA J Am Med Assoc 256:1141
Vroumsia T, Steiman R, Seigle-Murandi F, Benoit-Guyod JL (2005) Fungal bioconversion of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4-dichlorophenol (2,4-DCP). Chemosphere 60:1471–1480
del Pilar Castillo M, Andersson A, Ander P, StenstroÈm J, Torstensson L (2001) Establishment of the white rot fungus Phanerochaete chrysosporium on unsterile straw in solid substrate fermentation systems intended for degradation of pesticides. World J Microbiol Biotechnol 17:627–633
Cederlund H, Borjesson E, Onneby K, Stenstrom J (2007) Metabolic and cometabolic degradation of herbicides in the fine material of railway ballast. Soil Biol Biochem 39:473–484
Cabrera MI, Martín CA, Alfano OM, Cassano AE (1997) Photochemical decomposition of 2,4-dichlorophenoxyacetic acid (2,4-D) in aqueous solution. I. Kinetic study. Water Sci Technol 35:31–39
Beltran F, Gonzalez M, Rivas F, Jaramillo J (1995) Application of photochemical reactor models to UV irradiation of trichloroethylene in water. Chemosphere 31:2873–2885
Zertal A, Sehili T, Boule P (2001) Photochemical behaviour of 4-chloro-2-methylphenoxyacetic acid: influence of pH and irradiation wavelength. J Photochem Photobiol A 146:37–48
Zertal A, Jacquet M, Lavedrine B, Sehili T (2005) Photodegradation of chlorinated pesticides dispersed on sand. Chemosphere 58:1431–1437
Singh H, Saquib M, Haque MM, Muneer M (2007) Heterogeneous photocatalysed degradation of 4-chlorophenoxyacetic acid in aqueous suspensions. J Hazard Mater 142:374–380
Ranjit KT, Willner I, Bossmann S, Braun A (1998) Iron(III) phthalocyanine-modified titanium dioxide: a novel photocatalyst for the enhanced photodegradation of organic pollutants. J Phys Chem B 102:9397–9403
Modestov A, Glezer V, Marjasin I, Lev O (1997) Photocatalytic degradation of chlorinated phenoxyacetic acids by a new buoyant titania-exfoliated graphite composite photocatalyst. J Phys Chem B 101:4623–4629
Peller J, Wiest O, Kamat PV (2003) Synergy of combining sonolysis and photocatalysis in the degradation and mineralization of chlorinated aromatic compounds. Environ Sci Technol 37:1926–1932
Djebbar K, Sehili T (1998) Kinetics of heterogeneous photocatalytic decomposition of 2,4-dichlorophenoxyacetic acid over titanium dioxide and zinc oxide in aqueous solution. Pestic Sci 54:269–276
Sojic DV, Despotovic VN, Abazovic ND, Comor MI, Abramovic BF (2010) Photocatalytic degradation of selected herbicides in aqueous suspensions of doped titania under visible light irradiation. J Hazard Mater 179:49–56
Modestov A, Lev O (1998) Photocatalytic oxidation of 2,4-dichlorophenoxyacetic acid with titania photocatalyst. Comparison of supported and suspended TiO2. J Photochem Photobiol A 112:261–270
Abramovic BF, Sojic DV, Anderluh VB, Abazovic ND, Comor MI (2009) Nitrogen-doped TiO2 suspensions in photocatalytic degradation of mecoprop and (4-chloro-2-methylphenoxy)acetic acid herbicides using various light sources. Desalination 244:293–302
Flox C, Cabot PL, Centellas F, Garrido JA, Rodríguez RM, Arias C, Brillas E (2006) Electrochemical combustion of herbicide mecoprop in aqueous medium using a flow reactor with a boron-doped diamond anode. Chemosphere 64:892–902
Boye B, Brillas E, Dieng MM (2003) Electrochemical degradation of the herbicide 4-chloro-2-methylphenoxyacetic acid in aqueous medium by peroxi-coagulation and photoperoxi-coagulation. J Electroanal Chem 540:25–34
Brillas E, Boye B, Baños MÁ, Calpe JC, Garrido JA (2003) Electrochemical degradation of chlorophenoxy and chlorobenzoic herbicides in acidic aqueous medium by the peroxi-coagulation method. Chemosphere 51:227–235
Boye B, Brillas E, Marselli B, Michaud P-A, Comninellis C, Farnia G, Sandonà G (2006) Electrochemical incineration of chloromethylphenoxy herbicides in acid medium by anodic oxidation with boron-doped diamond electrode. Electrochim Acta 51:2872–2880
Lang K, Luňák S (2002) Photocatalytic degradation of 4-chlorophenoxyacetic acid in the presence of an iron complex and hydrogen peroxide. Photoch Photobio Sci 1:588–591
Zertal A, Molnár-Gábor D, Malouki M, Sehili T, Boule P (2004) Photocatalytic transformation of 4-chloro-2-methylphenoxyacetic acid (MCPA) on several kinds of TiO2. Appl Catal B 49:83–89
Ren X, Sun Y, Wu Z, Meng F, Cui Z (2012) The OH-induced degradation mechanism of 4-chloro-2-methylphenoxyacetic acid (MCPA) with two forms in the water: a DFT comparison. Chemosphere 88:39–48
Zhao Y, Truhlar DG (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J Phys Chem A 108:6908–6918
Frisch MJ (2004) GAUSSIAN 03, revision D.01. Gaussian Inc., Wallingford
Xu F, Wang H, Zhang Q, Zhang R, Qu X, Wang W (2010) Kinetic properties for the complete series reactions of chlorophenols with OH radicals—relevance for dioxin formation. Environ Sci Technol 44:1399–1404
Zhang Q, Yu W, Zhang R, Zhou Q, Gao R, Wang W (2010) Quantum chemical and kinetic study on dioxin formation from the 2,4,6-TCP and 2,4-DCP precursors. Environ Sci Technol 44:3395–3403
Zhang Q, Li S, Qu X, Shi X, Wang W (2008) A quantum mechanical study on the formation of PCDD/Fs from 2-chlorophenol as precursor. Environ Sci Technol 42:7301–7308
Fukui K (1981) The path of chemical reactions—the IRC approach. Acc Chem Res 14:363–368
Gonzalez C, Schlegel H (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Han Z, Zhang D, Sun Y, Liu C (2009) Reexamination of the reaction of 4-chlorophenol with hydroxyl radical. Chem Phys Lett 474:62–66
Kılıç M, Çınar Z (2008) Hydroxyl radical reactions with 4-chlorophenol as a model for heterogeneous photocatalysis. J Mol Struct (THEOCHEM) 851:263–270
Lukes P, Locke BR (2005) Degradation of substituted phenols in a hybrid gas–liquid electrical discharge reactor. Ind Eng Chem Res 44:2921–2930
Topalov AS, Sojic DV, Molnár-Gábor DA, Abramovic BF, Comor MI (2004) Photocatalytic activity of synthesized nanosized TiO2 towards the degradation of herbicide mecoprop. Appl Catal B 54:125–133
Devi KJ, Parveen S, Chandra AK (2009) Theoretical studies on the kinetics and thermochemistry of the gas-phase addition and H-abstraction reactions of 4-picoline with OH radical. Chem Phys Lett 480:161–167
Zertal A, Jacquet M, Lavérine B, Sehili T (2005) Photodegradation of chlorinated pesticides dispersed on sand. Chemosphere 58:1431–1437
Ennaceur S, Gandoura N, Driss M (2008) Distribution of polychlorinated biphenyls and organochlorine pesticides in human breast milk from various locations in Tunisia: levels of contamination, influencing factors, and infant risk assessment. Environ Res 108:86–93
Iwata H, Tanabe S, Sakai N, Tatsukawa R (1993) Distribution of persistent organochlorines in the oceanic air and surface seawater and the role of ocean on their global transport and fate. Environ Sci Technol 27:1080–1098
Qamar M, Muneer M, Bahnemann D (2006) Heterogeneous photocatalysed degradation of two selected pesticide derivatives, triclopyr and daminozid in aqueous suspensions of titanium dioxide. J Environ Manage 80:99–106
Konstantinou IK, Sakkas VA, Albanis TA (2002) Photocatalytic degradation of propachlor in aqueous TiO2 suspensions. Determination of the reaction pathway and identification of intermediate products by various analytical methods. Water Res 36:2733–2742
Liu N, Cui H, Yao D (2009) Decomposition and oxidation of sodium 3,5,6-trichloropyridin-2-ol in sub- and supercritical water. Process Saf Environ Prot 87:387–394
Topalov A, Molnár-Gábor D, Kosanic M, Abramovic B (2000) Photomineralization of the herbicide mecoprop dissolved in water sensitized by TiO2. Water Res 34:1473–1478
Acknowledgments
This work was supported by a project of the Shandong Province Science and Technology Department (no. 2010177) and a project of the Shandong Province Higher Educational Science and Technology Program (no. J09LB08). We also thank the China Postdoctoral Science Foundation (no. 20090461215).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1651 kb)
Rights and permissions
About this article
Cite this article
Ren, X., Sun, Y., Fu, X. et al. DFT comparison of the OH-initiated degradation mechanisms for five chlorophenoxy herbicides. J Mol Model 19, 2249–2263 (2013). https://doi.org/10.1007/s00894-013-1760-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-013-1760-9