
Abstract
Diuron is a phenyl urea herbicide used to control weeds in agricultural lands. The degradation of diuron in the atmosphere takes place dominantly via reaction with OH radicals. In this work, the OH addition reaction of diuron has been studied by using density functional theory methods M06-2X, ωB97X-D and MPWB1K with 6-31G(d,p) basis set. The calculated thermochemical parameters show that OH addition reaction occurs favourably at C2 position of diuron. The rate constant is calculated for the favourable initial reaction pathway by using canonical variational transition state theory with small curvature tunnelling (SCT) correction over the temperature range of 200–1000 K. The reaction of initially formed diuron-OH adduct intermediate with O2 leads to the formation of peroxy radical intermediate. The reaction of peroxy radical intermediate with HO2 and NOx (x = 1, 2) radicals is studied in detail. The results obtained from time-dependent density functional theory (TDDFT) calculations show that the intermediates and products formed from oxidation of diuron can be easily photolyzed in the sunlight. This study provides thermodynamical and kinetic data for the atmospheric oxidation of diuron by OH radical addition reaction and demonstrates the atmospheric chemistry of diuron and its derivatives.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenylurea herbicides are used to control unwanted weeds, insects, bacteria and fungi in the agricultural lands (Chang et al. 2016; Remucal 2014). Phenylurea herbicides exhibit high lipophilicity, bioaccumulation, long half-life and long range transporting ability which increase the chances of contaminating air, soil and water, even after many years of application (Jayaraj et al. 2016). The poisoning of environment due to pesticides is mainly by anthropogenic activity. Pesticides enter into the atmosphere by vapour shift during application, wind erosion from treated plants and soil and post-application volatization from surface of treated plants and soil (Chiron et al. 2000; Glotfelty 1978; Planas et al. 1997; Tomlin 2009). Globally, phenylurea herbicides have been detected in surface water and ground water in the areas where it is extensively used (Fingler et al. 2017). Various technologies have been emerged in recent years to remove the pesticides from the environment, such as dry and wet deposition (Waite et al. 1999), advanced oxidation process, adsorption filters and biological treatments (Malato et al. 2003). However, all these techniques do not completely remove the pesticides from the environment. Previous studies reported that abiotic transformation effectively removes the pesticides from the environment and their by-products often turned out to be hazardous than the parent pesticide (Hussain et al. 2015).
One of the commercially used phenylurea herbicides is diuron (3-(3, 4-dichloro-phenyl)-1,1-dimethyl urea) and is used for herbaceous, perennial weed control and pre-emergent treatment of fruit crops (Zhang et al. 2018). Diuron is also used in antifouling paints (Hussain et al. 2015) and is used as soil sterilant in agricultural lands (Polcaro et al. 2004). Diuron has high level of phytotoxicity and act in wide variety of plants as photosynthetic inhibitors (Guzzella et al. 2006). Diuron is used worldwide for more than 40 years and the total world production is estimated to be 14,000 to 16,000 tons per year (Esposito et al. 1998). Diuron is suspected to be carcinogenic, mutagenic, teratogenic, endocrine disrupting and genotoxic effects on animals as well as human (Krieger 2010; Remucal 2014). Pimentel (1995) reported that only a small percentage (0.3%) of applied pesticides go into target pest and the remaining 99.7% enter into the environment. Atmospheric fate of volatile organic compounds that are emitted into the atmosphere is mainly determined by their reactions with OH radical. Depending on the organic molecule, the reaction starts with either H-atom abstraction from organic molecule by OH radical or OH addition at relevant site of organic molecule.
Earlier experiments by Canle López et al. (2005) studied the reaction of diuron with OH radical using TiO2-photocatalyzed UV degradation technique and reported the rate constant at 298 K as 48.2 × 10−13 cm3 molecule−1 s−1. Kovács et al. (2015) used the pulse radiolysis technique to study the same reaction and the rate constant is determined as 96.4 × 10−13 cm3 molecule−1 s−1. Theoretically, Ren et al. (2014) studied the initial OH addition reaction of diuron in aqueous medium and reported that OH addition at C2 position of diuron is more favourable. Recently, we have studied the diuron oxidation through hydrogen atom abstraction reaction by OH radical (Manonmani et al. 2019) and found that the end-products formed from the secondary reactions are equally toxic as the parent diuron. Experimentally, the rate constant for the reaction of diuron with OH radical has been reported only at room temperature. Detailed kinetics and subsequent reactions for OH addition reaction of diuron have not yet been studied. With this exposure, the objective of the study is to characterize the potential energy surface for OH addition reaction of diuron by mapping out the various possible reaction pathways using computational methods. The subsequent reactions of diuron-OH adduct intermediate formed from favourable initial OH radical addition reaction with O2, HO2 and NOx (x = 1, 2) are studied in detail and various end-products were identified. The calculated rate constant for the favourable initial reactions over the temperature of 200–1000 K is used to study the competences of OH radical addition and hydrogen atom abstraction reactions. Furthermore, the theoretical insights on reaction mechanism for OH addition reaction of diuron and the products formed from the subsequent reactions allowed us to explore the atmospheric impacts of diuron emission.
Computational details
The geometry of reactant, transition states, intermediates and products involved in the reaction of diuron and OH radical was optimized by using density functional theory methods, M06-2X (Zhao and Truhlar 2008), ωB97X-D (Chai and Head-Gordon 2008) and MPWB1K (Zhao and Truhlar 2004) with 6-31G(d,p) basis set. Single-point energy calculations were performed at M06-2X/aug-cc-pVDZ//M06-2X/6-31G(d,p) level of theory. The highly accurate CBS-QB3 (Montgomery Jr et al. 2000) method was used to calculate the thermochemical parameters, such as relative enthalpy (∆H298) and Gibbs free energy (∆G298). The vibrational frequency calculations have been carried out at the above level of theories to verify the nature of stationary points. The transition state had single imaginary frequency, and intermediates and products have all positive frequencies. Synchronous transit- guided quasi-Newton (STQN) method is an efficient method to locate the transition state (Peng et al. 1996). This method uses linear synchronous transit (LST) or quadratic synchronous transit (QST) approach to get closer to the quadratic region of the transition state. In the QST framework, there are two methods to locate the transition state; QST2 method requires two molecular specifications, the reactant and product as the input. QST2 generates a guess for the transition state structure which is the midway between reactant and product, and then it optimize the guessed structure for the first order saddle point. Another method, QST3, requires three molecular specifications, reactant, product and guessed transition state structure (TSs) as its input. Then, the guessed TSs will be optimized. In the present study, for most of the studied pathways, TSs are located by using QST3 method and in few cases, QST2 method is used. In order to assure that the predicted transition state properly connects the designated reactant and product, the intrinsic reaction coordinate (IRC) (Fukui 1997, Gonzalez and Schlegel 1989, 1990) calculation has been performed at the same level of theories. In order to get accurate energies, a tight convergence criterion of 10−6 is used to optimize the structure of all the reactive species involved in the studied reactions. For the reactive species involved in the studied reactions, spin contamination (S2) is greater than or equal to 0.75 before annihilation which is greater than or equal to the optimal value of 0.75 after projection. That is, the spin contamination is not significant in the studied reactions. Previous studies have shown that solvation model density (SMD) continuum model is good enough to describe the solvent effect on energies of reactive species and is computationally less demanding than the other continuum models (Kabanda and Serobatse 2018; Marenich et al. 2009; Ponnusamy et al. 2017b). In the present study, the solvation model density (SMD) continuum model (Marenich et al. 2009) is employed to study the initial OH addition reaction of diuron. All the quantum chemical calculations were performed using GAUSSIAN 09 program package (Frisch et al. 1971). For the favourable initial reaction pathways, the rate constant is calculated using canonical variational transition state theory with small curvature tunnelling correction over the temperature range of 200–1000 K using GAUSSRATE 2016 program (Zheng et al. 2016b) which is an interface program between GAUSSIAN 09 and POLYRATE 2016 programs (Zheng et al. 2016a).
Results and discussion
Initial reactions
For diuron oxidation, as shown in Scheme 1, six possible pathways, R1–R6, were identified for OH addition reaction, depending on the position where the OH radical is attached on the carbon atom of diuron molecule. As given in Table S2, the energy of the reactive species optimized with different DFT methods is similar with a maximum deviation of 3 kcal/mol. Further, it has been observed that the structure of the reactive species optimized at different levels of theories is similar, and the relative importance of particular reaction path is the same in all studied methods. The results obtained from single-point energy calculation performed at M06-2X/aug-cc-pVDZ//M06-2X/6-31G(d,p) level of theory are comparable with that of other methods. The meta-hybrid exchange and correlation functional, M06-2X, is the most widely used method for studying the chemical reactions since it provides reliable results to study reaction mechanism and kinetics (Baruah et al. 2018; Ponnusamy et al. 2017a; Rao and Gejji, 2017; Sandhiya and Senthilkumar 2014). Therefore, the structure and energetics obtained from M06-2X functional are used to discuss reaction mechanism and rate constant calculations. The relative energy profile for the initial OH addition reaction of diuron calculated at M06-2X/6-31G(d,p) level of theory is shown in Fig. 1. The optimized geometry of the reactive species involved in the initial OH addition reaction is shown in Figs. 2 and S1. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) involved in the initial OH addition reaction of diuron are summarized in Table S2. As the reactants diuron and OH radical approach each other, the reactant complexes, RC1–RC4, were formed which are more stable than that of the isolated reactants by 5 to 10 kcal/mol. As shown in Fig. S1, RC1–RC4 were formed due to hydrogen bonding and Van der Waals interaction between diuron and OH radical.

The reaction scheme corresponding to the initial OH addition reaction of diuron

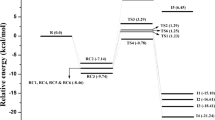
The relative energy profile corresponding to the initial OH addition reaction of diuron calculated at M06-2X/6-31G(d,p) level of theory

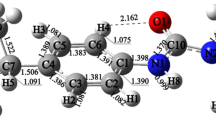
The optimized geometry of the transition states involved in the OH addition reaction of diuron at M06-2X/6-31G(d,p) level of theory. The selected bond distances are in angstrom
As shown in Scheme 1 and Figs. 2 and S1, pathways R1 and R2 correspond to the OH addition at C5 and C6 carbon atoms through the reactant complex RC1 and the transition states TS1 and TS2 which lead to the formation of the diuron-OH adduct intermediates I1 and I2. In the transition states, TS1 and TS2, the distance between carbon atom (C5/C6) of diuron and oxygen (O2) atom of OH radical decreases by 1.5 and 0.89 Å, respectively, from that of the reactant complex RC1. The energy barriers associated with the reactions R1 and R2 are 11.32 and 7.58 kcal/mol, respectively, and the reaction enthalpy (∆H298) and Gibbs free energy (∆G298) are − 18.21 and − 8.69 kcal/mol for I1 and − 24.19 and − 13.28 kcal/mol for I2; that is, the reactions are exothermic and exoergic in nature.
The pathway R3 corresponds to OH addition at C1 carbon atom which leads to the formation of the OH adduct intermediate I3 with an energy barrier of 9.95 kcal/mol. The intermediate I3 is formed through the reactant complex RC2 and the transition state TS3. In the transition state TS3, the distance between C1 and O2 atoms is 1.98 Å which is 0.85 Å shorter than that of the reactant complex RC2. The ∆H298 and ∆G298 for the formation of the diuron-OH adduct intermediate, I3, are − 23.12 and − 20.55 kcal/mol, respectively. The next pathway, R4, corresponds to OH addition at C2 carbon atom through the reactant complex, RC3, and the transition state, TS4. In the transition state, TS4, the distance between C2 and O2 atoms is decreased by 1.04 Å from that of the reactant complex, RC3. The energy barrier associated with the formation of diuron-OH adduct intermediate, I4, is 4.80 kcal/mol and the reaction is exothermic and exoergic by − 25.09 and − 14.78 kcal/mol, respectively. As shown in Scheme 1, pathways R5 and R6 correspond to OH addition at C3 and C4 carbon atoms which leads to the formation of products, 3-(4-chloro-3-hydroxy-phenyl)-1, 1-dimethyl urea (P1) and 3-(3-chloro-4-hydroxy-phenyl)-1, 1-dimethyl urea (P2) by eliminating the chlorine atom through the transition states, TS5 and TS6. In the transition states, TS5 and TS6, the distance between carbon atom, C3/C4, and oxygen atom (O2) is decreased by 0.84 Å from that of the reactant complex, RC4. The energy barriers associated with the formation of products, P1 and P2, are 9.35 and 7.98 kcal/mol, respectively. The relative enthalpy (∆H298) and Gibbs free energy (∆G298) for the formation of P1 are − 32.26 and − 22.71 kcal/mol and for P2 are − 32.31 and − 23.06 kcal/mol, respectively. Experimentally, Tanaka et al. (1986) and Jirkovský et al. (1997) identified the products, P1 and P2, in aqueous solution from phototransformation of diuron in natural sunlight and UV light and reported that the products are highly toxic. Previously, Ren et al. (2014) studied the OH addition reaction of diuron at MPWB1K/6–31 + G(d,p) level of theory in aqueous medium and reported that the energy barrier for the reaction pathways, R1–R6, is 11.01, 8.12, 12.49, 1.44, 12.84 and 10.10 kcal/mol, respectively, which is differed by ~ 3 kcal/mol with respect to the present gas phase results performed at M06-2X/6-31G(d,p), ωB97X-D/6-31G(d,p) and MPWB1K/6-31G(d,p) level of theories. In order to compare the gas and aqueous phase results, we studied the initial OH addition reaction in aqueous phase at M06-2X/6-31G(d,p) level of theory using solvation model density (SMD) continuum model and the results are summarized in Table S3. From Tables S2 and S3, it is observed that the energy barrier and reaction enthalpy calculated from aqueous phase are differed by 1–3 kcal/mol compared with that of gas phase results, showing milder solvent effect on the studied reactions. Among the studied initial OH addition reactions, the energy barrier associated with the formation of the OH adduct intermediate, I4, is relatively low; that is, the formation of I4 is more favourable than the other intermediates and products. By comparing the ∆H298 and ∆G298 values, it is observed that the reaction pathways, R4–R6, are highly exothermic and exoergic. Therefore, the rate constant is calculated for the reaction pathways corresponding to the formation of the products P1 and P2 and the intermediate I4 (R4–R6).
The rate constant for the favourable initial reactions (R4–R6) are studied using canonical variation transition state theory (CVT) with small curvature tunnelling (SCT) correction over the temperature range of 200–1000 K and at 1-bar atmospheric pressure. The rate constants calculated from transition state theory (TST) and canonical variational transition state theory (CVT) with and without small curvature tunnelling (SCT) correction are summarized in Tables 1 and S4. From Tables 1 and S4, it is observed that variational and tunnelling effects on the rate constant are negligible over the temperature range (200–1000 K) studied. The Arrhenius plot shown in Fig. 3 shows the positive temperature dependence of the rate constant. The rate constant corresponding to the formation of the diuron-OH adduct intermediate I4 and the products P1 and P2 is strongly dependent on the temperature. At higher temperatures, ranging from 600 to 1000 K, the rate constant is in the order 10−14 to 10−13 and at lower temperature range, 200–550 K, the rate constant is in the order 10−18 to 10−15. At 298 K, the CVT/SCT rate constants for the formation of I4, P1 and P2 are 3.96 × 10−16, 2.07 × 10−18 and 2.70 × 10−17 cm3 molecule−1 s−1, respectively.

The Arrhenius plot for the CVT/SCT rate constant of initial reactions, R4–R6, over the temperature range of 200–1000 K
Experimentally, Canle López et al. (2005) studied the reaction of diuron with OH radical at 298 K and the reported rate constant is 48.2 × 10−13 cm3 molecule−1 s−1. In the above experimental study, the dominant product identified was 1-acetyl-3- (3,4-dichloro-phenyl)-1- methyl urea and is confirmed from our previous study on the hydrogen atom abstraction reaction of diuron initiated by OH radical (Manonmani et al. 2019). Though the calculated energy barrier for favourable H-atom abstraction and OH addition reactions are differed by 0.3 kcal/mol, the rate constant calculated for H-atom abstraction reaction is higher by 103 than that of OH addition reaction at 298 K. This is due to the fact that the Gibbs free energy calculated for favourable H-atom abstraction reaction is − 21.08 kcal/mol which is higher by ~ 6 kcal/mol than the favourable OH radical addition reaction (Manonmani et al. 2019). By comparing the rate constant calculated for OH addition and hydrogen atom abstraction reactions, it is observed that the OH addition reaction is a temperature-driven mechanism. At 1000 K, the calculated rate constants for the formation of I4, P1 and P2 are 1.09 × 10−13, 6.85 × 10−14 and 1.75 × 10−13 cm3 molecule−1 s−1, respectively.
In order to study the contribution of I4, P1 and P2 to the degradation of diuron, the branching ratios of ГI4, ГP1 and ГP2 are calculated and are summarized in Table 2. At 298 K, the branching ratios calculated for I4, P1 and P2 are 93%, 1% and 6%, respectively; that is, at room temperature, the formation of I4 is more favourable and at the temperature range of 200–800 K, the branching ratios calculated for products P2 and I4 are comparable. At a higher temperature ranging from 800 to 1000 K, the calculated branching ratio show that formation of P2 is more favourable. The broad energy barrier with imaginary frequency of − 628.68 cm−1 and high activation free energy of transition state, TS6 (14.34 kcal/mol), corresponding to the formation of P2 is the reason for the magnitude of the rate constant at high temperature. The products P1 and P2 were identified experimentally by Jirkovský et al. (1997) as important photoproducts obtained from photolysis of diuron. The above results show the possibility of formation of I4 and P2 from the initial OH radical addition reaction. Hence, the subsequent reactions associated with the intermediate I4 are important in the atmospheric chemistry of diuron.
Subsequent reactions of diuron-OH adduct intermediate, I4
Under atmospheric condition, it is important to study the reaction of diuron-OH adduct intermediate, I4, with O2. The scheme for the reactions of intermediate, I4, with O2 is shown in Scheme 2. The relative energy profile is shown in Fig. 4 and the optimized geometry of the stationary points on the reaction potential energy surface is shown in Figs. 5 and S2. The calculated relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) are summarized in Table S5.

The reaction scheme for reactions of intermediate, I4 with O2

The relative energy profile corresponding to the reaction of I4 with O2 calculated at M06-2X/6-31G(d,p) level of theory. The energy values given in the parentheses are in kilocalorie per mole

The optimized structure of transition states for the reactions of I4 with O2 at M06-2X/6-31G(d,p) level of theory. The selected bond distances are given in angstrom
As shown in Scheme 2, the O2 addition at C5, C4, C3, C2 and C6 positions of intermediate, I4, leads to the formation of peroxy radical intermediates, I5–I9, through the reactant complexes, RC5–RC8, and transition states, TS7–TS11, with energy barriers of 11.31, 43.29, 7.39, 8.60 and 29.39 kcal/mol, respectively. The reaction associated with the formation of peroxy radical intermediates, I5, I7 and I8, are highly exothermic with reaction enthalpy of − 44.02, − 46.18 and − 42.69 kcal/mol, respectively, and for intermediates, I6 and I9, the reaction is slightly exothermic by − 2.67 and − 1.73 kcal/mol, respectively. As shown in Scheme 2, the reaction of I4 with O2 leads to the formation of 3-(3,4-dichloro-2-hydroxy-phenyl)-1-1-dimethyl urea (P3) and HO2 radical through the transition state, TS12, with an energy barrier of 10.8 kcal/mol. In the transition state, TS12, the distance between H1 and O3 atoms is 1.58 Å which is 1.3 Å lower than that of the reactant complex, RC9. The formation of P3 is highly exothermic and exoergic by − 40.58 and − 39.72 kcal/mol, respectively. By comparing the energy barrier, ∆H298 and ∆G298 values for the reaction of I4 and O2, it is found that the formation of intermediates, I7 and I8, are equally important.
Note that previous studies (Gnanaprakasam et al. 2017; Shiroudi et al. 2014) reported that the unimolecular isomerization reaction for peroxy radical intermediates is found to be unlikely in the atmosphere due to high energy barrier. Therefore, in the present study, we have not studied the unimolecular isomerization reaction for intermediates I7 and I8. Further, the subsequent reactions of I7 and I8 with other atmospheric reactive species like HO2 and NO and dissociation of alkoxy radicals formed from I7 and I8 are studied and discussed in the following section.
Subsequent reactions of intermediate, I7
The reaction scheme for the subsequent reactions of intermediate, I7, is shown in Scheme 3. The relative energy profile and optimized structure of reactant complexes, transition states, intermediates and products are shown in Figs. 6 and 7 and S3. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) for the reactive species involved in the subsequent reactions of intermediate, I7, are summarized in Table S6.

The reaction scheme for the subsequent reactions of intermediate, I7

The relative energy profile (Gibbs free energy) of the subsequent reactions of intermediate, I7, calculated at M06-2X/6-31G(d,p) level of theory. The energy values given in the parentheses are in kilocalorie per mole

The optimized structure of the transition states involved in the subsequent reactions of intermediate, I12. The selected bond distances are given in angstrom
As shown in Scheme 3, the first possible exit pathway for I7 is its reaction with HO2 radical. In this reaction, the product, 3-(4,5-dichloro-5-hydroperoxy-6-hydroxy-cyclohexa-1,3-dienyl)-1, 1-dimethyl urea, P4 and O2 are formed through the transition state, TS13, with an energy barrier of 2.52 kcal/mol. In TS13, the distance between H11 and O4 atoms is 1.88 Å which is 0.7 Å smaller than that of the reactant complex, RC10. The relative enthalpy (∆H298) and Gibbs free energy (∆G298) for the corresponding reaction are − 27.74 and − 30.51 kcal/mol, respectively. The next possible pathway for intermediate, I7, is its reaction with NO radical which leads to the formation of peroxy nitrite intermediate, I10, in a barrierless process. This reaction is highly exothermic and exoergic by − 34.73 and − 34.60 kcal/mol, respectively. As shown in Scheme 3, the peroxy nitrite intermediate, I10, can transform in to alkoxy radical intermediate, I11, by eliminating NO2 radical in a barrierless reaction. The formation of alkoxy radical intermediate, I11, is slightly endothermic and the reaction is barrierless which is in agreement with our previous study (Gnanaprakasam et al. 2018). As reported in previous studies (Gnanaprakasam et al. 2018; Vereecken and Francisco 2012), the alkoxy radical can undergo decomposition and oxidation reaction. Therefore, from the alkoxy radical intermediate, I14, three possible reaction pathways are studied which results in the formation of stable products, P5-P7. In the first pathway, I11 reacts with HO2 radical resulting in the formation of 3-(4, 5-dichloro-5,6-dihydroxy-cyclohexa-1,3-dienyl)-1,1-dimethyl urea, P5 and O2 through the transition state, TS14, with an energy barrier of 2.47 kcal/mol. In the transition state, TS14 the distance between H12 and O3 atoms is 1.63 Å which is 0.3 Å smaller than that of I11 + HO2. The formation of P5 is highly exothermic and exoergic by − 44.12 and − 47.21 kcal/mol, respectively. In the next possible pathway, the product, P6, is formed with the elimination of chlorine atom (Cl2) through the transition state, TS15, with an energy barrier of 0.56 kcal/mol. In TS15, the distance between C3 and Cl2 atoms is 1.98 Å which is 0.2 Å smaller than that of intermediate, I11. The corresponding reaction is exothermic with ∆H298 of − 25.90 kcal/mol and exoergic with ∆G298 of − 27.39 kcal/mol. Another possible exit pathway for the intermediate, I11, is the formation of 3-(4, 5-dichloro-5,6-dihydroxy-4-methyl-cyclohex-1-enyl)-1,1-dimethyl urea, P7, through the transition state, TS16, for which the energy barrier is 1 kcal/mol. In the transition state, TS16, the distance between C4 and O3 atoms is 1.86 Å which is 0.23 Å smaller than that of intermediate, I11. The reaction corresponding to the formation of product, P7, is slightly exothermic and exoergic by − 9.16 and − 8.98 kcal/mol, respectively. By comparing the energy barrier, enthalpy and Gibbs free energy for the subsequent reactions of I4, it is found that the reactions corresponding to the formation of products, P4–P7, are equally favourable.
Subsequent reactions of intermediate, I8
The reaction scheme for the subsequent reactions of intermediate, I8, is shown in Scheme 4 and the corresponding relative energy profile is shown in Fig. 8. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) are summarized in Table S7. The optimized structure of the reactive species involved in the subsequent reactions of intermediate, I8, is shown in Figs. 9 and S4.

The reaction scheme for the subsequent reactions of the intermediate, I8

The relative energy profile of the secondary reactions of intermediate, I8, calculated at M06-2X/6-31G(d,p) level of theory. The energy values given in the parentheses are in kilocalorie per mole

The optimized structure of the transition states involved in the subsequent reactions of intermediate, I8. The selected bond distances are given in angstrom
As shown in Scheme 4, the reaction of I8 with HO2 radical leads to the formation of 3-(4, 5-dichloro-1-hydroperoxy-6-hydroxy-cyclohexa-2, 4-dienyl)-1-dimethyl urea (P8) and O2 through transition state, TS17, with an energy barrier of 4.88 kcal/mol. In the transition state, TS17, the distance between O4 and H12 atoms is 1.67 Å which is 0.4 Å smaller than that of reactant, I8 + HO2. The reaction associated with the formation of product, P8, is exothermic with an ∆H298 value of − 23.68 kcal/mol and exoergic with ∆G298 of − 27.18 kcal/mol. The peroxy radical intermediate, I8, can react with NO radical. As shown in Fig. 8 and Table S7, this reaction is barrierless and highly exothermic by − 33.33 kcal/mol. As reported in the previous study (Gnanaprakasam et al. 2018), the intermediate, I12, can dissociate into alkoxy radical intermediate, I13, and NO2 in a barrierless reaction. This reaction is endothermic and endoergic by 13.49 and 0.79 kcal/mol, respectively. As discussed earlier, the alkoxy radical undergoes oxidation reaction with HO2 which leads to the formation of 3-(4,5-dichloro-1,6-dihydroxy-cyclohexa-2,4-dienyl)-1,1-dimethyl urea, P9 and O2 through the transition state, TS18, with an energy barrier of 1.62 kcal/mol. In TS18, the distance between H12 and O3 atoms is 1.75 Å which is 0.3 Å shorter than that of reactant, I12 + HO2. The corresponding ∆H298 and ∆G298 are −47.17 and − 50.04 kcal/mol, respectively. That is, the reaction is highly exothermic and exoergic in nature. The next possible pathway from intermediate, I13, is the formation of 3-(3,4-dichloro-2-hydroxy-7-oxa-bicyclohept-3-enyl)-1,1-dimethyl urea, P10, through the transition state, TS19, with an energy barrier of 2.73 kcal/mol and the reaction is exothermic by − 12.49 kcal/mol. By comparing ∆E, ∆H298 and ∆G298 values for the subsequent reactions of the intermediate, I8, it is found that the reactions corresponding to the formation of products, P8–P10, are feasible.
Photolysis of diuron
Many research groups (Amine-Khodja et al. 2004; Jirkovský et al. 1997; Mazellier et al. 1997) have studied the photolysis of diuron in aqueous medium using experimental techniques and reported that the formation of photoproducts are more toxic than the parent diuron. However, in literature, there is no information on photolysis of diuron under typical environmental condition. In order to study photolysis of diuron in gas phase, the vertical excitation energy of isolated diuron, intermediates I1–I4 and products P1 and P2 formed from the initial reaction of diuron is calculated by using a time-dependent density functional theory (Gross et al. 1996) method at M06-2X/6-31G(d,p) level of theory and is summarized in Table 2, and the simulated absorption spectra is shown in Fig. 10. The dominant absorption spectra with oscillator strength higher than 0.05 are noted for intermediates I1–I4 and products P1 and P2. Bai et al. (Bai et al. 2015) reported that if the vertical excitation energy is lesser than 4.13 eV (~300 nm), the molecule will undergo photolysis in sunlight. As shown in Fig. 10, the dominant absorption for diuron is 220 nm and is in agreement with the experimental value of 214 nm (Djebbar et al. 2003). From Table S6, it is observed that photolysis of isolated diuron is not possible, because the excitation energy is greater than the threshold value of 4.13 eV. In the case of intermediates and products, the excitation energy is lesser than tropospheric cut-off 4.13 eV (~300 nm) which indicates the possibility of photolysis of intermediates and products. The intermediates I1–I4 can easily photolyze at about 303, 347, 307 and 351 nm, respectively. The products P1 and P2 can photolyze at about 518 and 573 nm. Note that these products were identified experimentally by Jirkovský et al. (1997). Hence, these intermediates and products are responsible for the formation of secondary organic aerosols in the atmosphere due to oxidation of diuron by OH radical.

The absorption spectra of a isolated diuron and b intermediates and products formed from the initial reaction of diuron with OH radical computed at TD-M06-2X/6-31G(d,p) level of theory
The products formed from the initial and subsequent reactions have profound implications in the atmosphere. As discussed earlier, the initially formed diuron-OH adduct intermediate I4 can react with O2 in the atmosphere, which leads to the formation of peroxy radical intermediates and 3-(3,4-dichloro-2-hydroxy-phenyl)-1-1-dimethyl urea, P3. The formed peroxy radical intermediate has a potential contribution in secondary organic aerosol formation and it slows down the free radical–driven chemical reactions which load the tropospheric ozone. As discussed earlier, the peroxy radical intermediates I7 and I8 are highly reactive and undergo reaction with HO2 and NO radicals. In the region with low NOx (where x = 1, 2) concentration, the reaction of HO2 with I7 and I8 dominates and results in the formation of 3-(4,5-dichloro-5-hydroperoxy-6-hydroxy-cyclohexa-1,3-dienyl)-1,1-dimethyl urea (P4) and 3-(4,5-dichloro-1-hydroperoxy-6-cyclohexa-2,4-dienyl)-1,1-dimethyl urea (P8) and O2 as a by-product. On the other hand, at the region where NOx (where x = 1, 2) concentration is high, the peroxy radicals I7 and I8 react with NO radical leading to the formation of products like 3-(4,5-dichloro-5,6-dihydroxy-cyclohexa-1, 3-dienyl)-1,1-dimethyl urea (P5), 3-(4-chloro-6-hydroxy-5-oxo-cyclohexa-1,3-dienyl)-1,1-dimethyl urea (P6), 3-(4, 5-dichloro-5,6-dihydroxy-4-methyl-cyclohex-1-enyl)-1,1-dimethyl urea (P7), 3-(4,5-dichloro-1,6-dihydroxy-cyclohexa-2,4-dienyl)-1,1-dimethyl urea (P9) and 3-(3,4-dichloro-2-hydroxy-7-oxa-bicyclo-hept-3-en-1-yl)-1,1-dimethyl urea (P10) with the elimination of O2, NO2 and Cl radicals which cause adverse effects in human and environment. Note that the reported reactions associated with the formation of products are highly exothermic and exoergic in nature. The energetics calculated for the reactive species corresponding to the formation of products show the equal probability for the formation of these products. The products formed are highly toxic as the parent molecule, diuron. The results from the photolysis study also show that the intermediates and products are more reactive. Further, the reported results clearly demonstrate the potential role of hydroxyl radical on the degradation of diuron in the atmosphere. The proposed reaction mechanism provides further insights on thermochemistry and kinetics of atmospheric reaction of analogous species.
Conclusion
In the present work, reaction mechanism and kinetics of OH addition reaction of diuron is studied by using electronic structure calculations. For the initial reaction, six OH addition reaction pathways were studied. The result shows that OH addition of diuron occurs favourably at C2 position of diuron with an energy barrier of 4.80 kcal/mol. The calculated rate constant and branching ratio show that OH addition reaction is more dominant at higher temperature range (600–1000 K), and the formation of the diuron-OH adduct intermediate I4 and 3-(3-chloro-4-hydroxy-phenyl)-1,1-dimethyl urea (P2) are equally probable. The reaction of the diuron-OH adduct intermediate I4 with O2 leads to the formation of peroxy radical intermediate. The possible reactions of this peroxy radical intermediate with other atmospheric species, such as HO2 and NO radicals, were studied, and it was found that the products 3-(4,5-dichloro-5-hydroperoxy-6-hydroxy-cyclohexa-1,3-dienyl)-1,1-dimethyl urea (P4), 3-(4, 5-dichloro-5,6-dihydroxy-cyclohexa-1,3-dienyl)-1,1-dimethyl urea (P5), 3-(4-chloro-6-hydroxy-5-oxo-cyclohexa-1,3-dienyl)-1,1-dimethyl urea (P6), 3-(4,5-dichloro-5,6-dihydroxy-4-methyl-cyclohex-1-enyl)-1,1-dimethyl urea (P7), 3-(4,5-dichloro-1-hydroperoxy-6-cyclohexa-2,4-dienyl)-1,1-dimethyl urea (P8), 3-(4,5-dichloro-1,6-dihydroxy-cyclohexa-2,4-dienyl)-1,1-dimethyl urea (P9) and 3-(3,4-dichloro-2-hydroxy-7-oxa-bicyclo-hept-3-en-1-yl)-1,1-dimethyl urea (P10) are formed from secondary reactions of diuron. Time-dependent density functional theory calculations show that intermediates and products formed from the initial reactions are easily photolyzed by sunlight. The products formed from initial and subsequent reactions are highly toxic than the parent diuron. Hence, the use of diuron in agricultural lands should be limited.
References
Amine-Khodja A, Boulkamh A, Boule P (2004) Photochemical behaviour of phenylurea herbicides. Photochem Photobiol Sci 3:145–156
Bai F-Y, Wang X, Sun Y-Q, Pan X-M (2015) Atmospheric chemistry of alkyl iodides: theoretical studies on the mechanisms and kinetics of CH 3 I/C 2 H 5 I+ NO 3 reactions. RSC Adv 5:88087–88095
Baruah SD, Gour NK, Sarma PJ, Deka RC (2018) OH-initiated mechanistic pathways and kinetics of camphene and fate of product radical: a DFT approach. Environ Sci Pollut Res 25:2147–2156
Canle López M, Fernandez MI, Rodríguez S, Santaballa JA, Steenken S, Vulliet E (2005) Mechanisms of direct and TiO2-photocatalysed UV degradation of phenylurea herbicides. ChemPhysChem 6:2064–2074
Chai J-D, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Chang P-L, Hsieh M-M, Chiu T-C (2016) Recent advances in the determination of pesticides in environmental samples by capillary electrophoresis. Int J Environ Res Public Health 13:409
Chiron S, Fernandez-Alba A, Rodriguez A, Garcia-Calvo E (2000) Pesticide chemical oxidation: state-of-the-art. Water Res 34:366–377
Djebbar K, Sehili T, Mazellier P, De Laat J (2003) Phototransformation of diuron in aqueous solution by UV irradiation in the absence and in the presence of H2O2. Environ Technol 24:479–489
Esposito E, Paulillo SM, Manfio GP (1998) Biodegradation of the herbicide diuron in soil by indigenous actinomycetes. Chemosphere 37:541–548
Fingler S, Mendaš G, Dvoršćak M, Stipičević S, Vasilić Ž, Drevenkar V (2017) Herbicide micropollutants in surface, ground and drinking waters within and near the area of Zagreb, Croatia. Environ Sci Pollut Res 24:11017–11030
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson G (1971): Gaussian 09, Revision A. 02; Gaussian, Inc: Wallingford, CT, 2009. Google Scholar
Fukui K (1997) The path of chemical reactions—the IRC approach, Frontier orbitals and reaction paths: selected papers of Kenichi Fukui. World Scientific, pp. 471–476
Glotfelty DE (1978) The atmosphere as a sink for applied pesticides. J Air Pollut Control Assoc 28:917–921
Gnanaprakasam M, Sandhiya L, Senthilkumar K (2017) A theoretical investigation on the mechanism and kinetics of the gas-phase reaction of naphthalene with OH radical. Theor Chem Accounts 136:131
Gnanaprakasam M, Sandhiya L, Senthilkumar K (2018) Mechanism and kinetics of the oxidation of dimethyl carbonate by hydroxyl radical in the atmosphere. Environ Sci Pollut Res:1–11
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J Chem Phys 90:2154–2161
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Gross EK, Dobson J, Petersilka M (1996): Density functional theory of time-dependent phenomena, density functional theory II. Springer, pp. 81–172
Guzzella L, Capri E, Di Corcia A, Barra Caracciolo A, Giuliano G (2006) Fate of diuron and linuron in a field lysimeter experiment. J Environ Qual 35:312–323
Hussain S, Arshad M, Springael D, SøRensen SR, Bending GD, Devers-Lamrani M, Maqbool Z, Martin-Laurent F (2015) Abiotic and biotic processes governing the fate of phenylurea herbicides in soils: a review. Crit Rev Environ Sci Technol 45:1947–1998
Jayaraj R, Megha P, Sreedev P (2016) Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip Toxicol 9:90–100
Jirkovský J, Faure V, Boule P (1997) Photolysis of diuron. Pestic Sci 50:42–52
Kabanda MM, Serobatse KR (2018) A DFT study on the addition and abstraction reactions of thiourea with hydroxyl radical. J Sulfur Chem 39:23–46
Kovács K, He S, Mile V, Csay T, Takács E, Wojnárovits L (2015) Ionizing radiation induced degradation of diuron in dilute aqueous solution. Chem Cent J 9:21
Krieger R (2010) Hayes’ handbook of pesticide toxicology, 1. Academic press
Malato S, Cáceres J, Fernández-Alba A, Piedra L, Hernando M, Agüera A, Vial J (2003) Photocatalytic treatment of diuron by solar photocatalysis: evaluation of main intermediates and toxicity. Environ Sci Technol 37:2516–2524
Manonmani G, Sandhiya L, Senthilkumar K (2019) Mechanism and kinetics of diuron oxidation initiated by hydroxyl radical: hydrogen and chlorine atom abstraction reactions. J Phys Chem A 123:8954–8967
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Mazellier P, Jirkovsky J, Bolte M (1997) Degradation of diuron photoinduced by iron (III) in aqueous solution. Pestic Sci 49:259–267
Montgomery JA Jr, Frisch MJ, Ochterski JW, Petersson GA (2000) A complete basis set model chemistry VII. Use of the minimum population localization method. J Chem Phys 112:6532–6542
Peng C, Ayala PY, Schlegel HB, Frisch MJ (1996) Using redundant internal coordinates to optimize equilibrium geometries and transition states. J Comput Chem 17:49–56
Pimentel D (1995) Amounts of pesticides reaching target pests: environmental impacts and ethics. J Agric Environ Ethics 8:17–29
Planas C, Caixach J, Santos F, Rivera J (1997) Occurrence of pesticides in Spanish surface waters. Analysis by high resolution gas chromatography coupled to mass spectrometry. Chemosphere 34:2393–2406
Polcaro AM, Mascia M, Palmas S, Vacca A (2004) Electrochemical degradation of diuron and dichloroaniline at BDD electrode. Electrochim Acta 49:649–656
Ponnusamy S, Sandhiya L, Senthilkumar K (2017a) The atmospheric oxidation mechanism and kinetics of 1, 3, 5-trimethylbenzene initiated by OH radicals–a theoretical study. New J Chem 41:10259–10271
Ponnusamy S, Sandhiya L, Senthilkumar K (2017b) Reaction mechanism and kinetics of the degradation of terbacil initiated by OH radical-a theoretical study. Chem Phys
Rao PK, Gejji SP (2017) Kinetics and mechanistic investigations of atmospheric oxidation of HFO-1345fz by OH radical: insights from theory. J Phys Chem A 121:595–607
Remucal CK (2014) The role of indirect photochemical degradation in the environmental fate of pesticides: a review. Environ Sci: Process Impacts 16:628–653
Ren X, Cui Z, Sun Y (2014) Theoretical studies on degradation mechanism for OH-initiated reactions with diuron in water system. J Mol Model 20:2280
Sandhiya L, Senthilkumar K (2014) Reaction mechanism and kinetics of the degradation of bromoxynil initiated by OH radical. RSC Adv 4:7749–7759
Shiroudi A, Deleuze MS, Canneaux S (2014) Theoretical study of the oxidation mechanisms of naphthalene initiated by hydroxyl radicals: the OH-addition pathway. J Phys Chem A 118:4593–4610
Tanaka F, Hoffer B, Wien R (1986) Photolysis of 3-(3, 4-dichlorophenyl)-1, 1-dimethylurea (diuron) in dilute aqueous solution. Toxicol Environ Chem 11:261–269
Tomlin CD (2009) The pesticide manual: a world compendium. British Crop Production Council
Vereecken L, Francisco JS (2012) Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem Soc Rev 41:6259–6293
Waite DT, Cessna AJ, Gurprasad NP, Banner J (1999) A new sampler for collecting separate dry and wet atmospheric depositions of trace organic chemicals. Atmos Environ 33:1513–1523
Zhang F, Liu B, Liu G, Zhang Y, Wang J, Wang S (2018) Substructure-activity relationship studies on antibody recognition for phenylurea compounds using competitive immunoassay and computational chemistry. Sci Rep 8:3131
Zhao Y, Truhlar DG (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J Phys Chem A 108:6908–6918
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167
Zheng J, Bao J, Meana-Pañeda R, Zhang S, Lynch B, Corchado J, Chuang Y, Fast P, Hu W, Liu Y (2016a) Polyrate-version 2016-2A. University of Minnesota, Minneapolis
Zheng J, Zhang S, Corchado J, Chuang Y, Coitiño E, Ellingson B, Truhlar D (2016b) GAUSSRATE version 2016. University of Minnesota, Minneapolis
Funding
The authors received funding from UGC and the Department of Science and Technology (DST), India, to establish high-performance computing facility under the SAP and PURSE programs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Vítor Pais Vilar
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Cartesian coordinates of the transition state structures involved in the studied reactions calculated at M06-2X/6-31G(d,p) level of theory are summarized in Table S1. The calculated relative energy (∆E, kcal/mol), enthalpy (∆H298, kcal/mol) and Gibbs free energy (∆G298, kcal/mol) for the initial OH addition reaction of diuron are given in Table S2. Relative energy (∆E, kcal/mol), enthalpy (∆H, kcal/mol) and Gibbs free energy (∆G, kcal/mol) of the reactive species involved in the initial OH addition reactions of diuron by OH radical in aqueous medium obtained at M06-2X/6-31G(d,p) level of theory are given in Table S3. The rate constant for the initial reactions calculated using CVT, TST and TST (SCT) methods are summarized in Table S4. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) of the reactive species involved in the reaction of intermediate, I4 with O2 are given in Table S5. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) of all the reactive species involved in the subsequent reactions of intermediate, I7 are given in Table S6. The relative energy (∆E), enthalpy (∆H298) and Gibbs free energy (∆G298) of all the reactive species involved in the subsequent reactions of intermediate, I8 are given in Table S7. The optimized geometry of reactant complexes, intermediates and products involved in the OH addition reaction of diuron are shown in Fig. S1. The structure of reactant complexes, intermediates and product for the reaction of I4 with O2 are shown in Fig. S2. The optimized structure of intermediates, products involved in the subsequent reactions of intermediate, I12 are shown in Fig. S3. The optimized structure of intermediates and products involved in the subsequent reactions of intermediate, I8 are shown in Fig. S4. (DOCX 934 kb)
Rights and permissions
About this article

Cite this article
Manonmani, G., Sandhiya, L. & Senthilkumar, K. Mechanism and kinetics of diuron oxidation by hydroxyl radical addition reaction. Environ Sci Pollut Res 27, 12080–12095 (2020). https://doi.org/10.1007/s11356-020-07806-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-07806-4