
Abstract
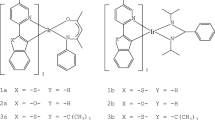
Iridium (III) 2-phenylpyridine (ppy) complexes with two suitable monodentate L ligands [Ir(ppy)2(L)2]+ (ppy = 2-phenylpyridine, py = pyridine, L = 4-pyCN 1, 4-pyCHO 2, 4-pyCl 3, py 4, 4-pyNH2 5) were studied by density functional theory (DFT) and time-dependent DFT methods. The influences of ligands L on the electronic structure and photophysical properties were investigated in detail. The compositions and energy levels of the lowest unoccupied molecular orbital (LUMO) are changed more significantly than those of the highest occupied molecular (HOMO) by tuning L ligands. With the electronegativity decrease of L ligands 4-pyCN > 4-pyCHO > 4-pyCl > py > 4-pyNH2, the LUMO distributing changes from py to ppy, and the absorptions have an obvious red shift. The calculated results showed that the transition character of the absorption and emission can be changed by adjusting the electronegativity of the L ligands. In addition, no solvent effect was observed in the absorptions and emissions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphorescent iridium (III) complexes have been explored for a variety of photonic applications including emitting diodes [1, 2], biological labeling [3], photosensitization [4, 5], and emissive materials [6] in electrochemiluminescent. Especially, six-coordinate cyclometallated Ir(III) complexes with one, two, or three 2-phenylpyridine (ppy), or derivative C^N ligands and other ancillary ligands [7–9], such as Ir(Mebib)(ppy)X (Mebib = bis(N-methylbenzimidazolyl)benzene; X = Cl, −C ≡ C, CN), Ir(η2-ppy)2(η2-XZY) (XZY = 2-mercaptopyridine (mp) and trimethylacetic acid (tma) etc.), Ir(ppy)3, and Ir(C^N)2LX (C^N = benzoquinoline (bzq), 2-(4-tolyl)pyridine (tpy), 2-(2΄-thienyl)pyridine (thp), LX = acetoylacetonate (acac), dibenzoylmethanate (dbm)), have received intensive attention due to their favorable photochemical and photophysical properties. The luminescence properties of Ir(III) complexes can be fine-tuned by ligand substituents, resulting in distinct emission color tuning [10].
On the other hand, Ir(III) complexes have also attracted considerable interest as sensor, because of their significant stokes shifts, long emissive lifetimes, and large emission shifts from change in the local environment compared with purely organic luminophores [11]. Although some Ir(III) compound-based chemosensors for anion [12–14] and oxygen [15, 16] have been described, the chemosensors to heavy metal ions are still less explored. To the best of our knowledge, Ir(btp)2(acac) (btp = 2-(benzo[b]thiophen-2-yl)pyridine) presents highly selective phosphorescent chemosensor for Hg2+, [Ir(ppy)2 L]PF6 (L = (4-[2,2΄]Bipyridinyl-5-ylethynylphenyl)pyridin-2-ylmethylamine) shows selective luminescence recognition to Zn2+, and [Ir(ppy)2L]PF6 (L = (4-[2,2΄]Bipyridinyl-5-ylethynylphenyl)thiophen-2-ylmethylamine) exhibits a unique off-on-off luminescence switching effect to Cu2+ [17, 18], which have been investigated experimentally and theoretically. Recently, a series of new cationic Ir(III) complexes [Ir(ppy)2L2]+ (L = 4-pyCHO, 4-pyNH2) have been synthesized, and X-ray crystal structures, absorption and emission spectra have been investigated by Sie et al. [19]. It is worth noting that the L ligand in Ir(III) complexes [Ir(ppy)2L2]+ is monodentate ligand, which is different from the bidentate L in Ir(C^N)2L complexes mentioned above. Meanwhile, the quantum yield of [Ir(ppy)2(4-pyNH2)2]+ is much higher than that of [Ir(ppy)2(4-pyCHO)2]+, and [Ir(ppy)2(4-pyNH2)2]+ has shown good chemical-sensing ability to silver salts. Therefore, by changing the substitutive group on the ancillary ligand (py) can change spectroscopic property and furthermore make change in potential application. But the theoretical study on the spectral properties of these complexes is sparse from an electronic structure point of view. In addition, the relationship between L ligands and the spectra in the complexes [Ir(ppy)2L2]+ is not clear. Therefore, a deep insight into the structures and spectroscopic properties for this kind of complexes is much needed and significant.
To foresee new structure-property relationships and reveal the effects of the ligands L on electronic structure and photophysical property, we carried out the quantum chemistry studies on the cationic Ir (III) 2-phenylpyridine complexes with two suitable monodentate ancillary ligands L, [Ir(ppy)2(L)2]+ (ppy = 2-phenylpyridine, py = pyridine, L = 4-pyCN 1, 4-pyCHO 2, 4-pyCl 3, py 4, 4-pyNH2 5), using density functional theory (DFT) methods [20]. In addition, the influences of different solvents on the spectra were also studied in detail. The present study presents useful information for the design of new phosphors based on [Ir(ppy)2(L)2]+ complexes with different ligands L in sensor.
Computation methods
All of the calculations were accomplished by using the Gaussian 03 software package [21]. A hybrid Hartree-Fock (HF)/DFT model approach based on the Perdew-Burke-Erzenrhof (PBE) functional [22, 23], referred to as PBE1PBE, where the HF/DFT exchange ratio is fixed a priori to 1/4, was used to optimize the ground state, and the unrestricted PBE1PBE (UPBE1PBE) method was used to optimize the excited state geometries. There were no symmetry constraints on these complexes. Although the high electron multiplicity is considered, the calculated spin contamination is rather small: the expectation values of spin operator <S 2> are about 2.03 for triplet states. Base on optimized geometries in the ground and the excited state, the molecular orbital compositions, and absorption and emission spectra in CH2Cl2 media were calculated by time-dependent DFT (TD-DFT) method [24–26] at the PBE1PBE hybrid functional level associated with the polarized continuum model (PCM) [27, 28]. In addition, the molecular orbital compositions (population analysis) were calculated by C-squared population analysis (SCPA) method [29] and the absorption spectra were simulated by means of Swizard [30] program using Gaussian functions with half-width of 3000 cm−1. In the calculations, the quasi-relativistic pseudo-potentials of Ir atom proposed by Hay and Wadt [31, 32] with 17 valence electrons were used. The LANL2DZ basis set associated with the pseudo-potential was adopted for Ir atom, and 6-31G(d) basis bet was adopted for other atoms. This kind of theoretical approach and calculation level has been proven to be reliable for transition-metal complex systems [33–37].
To explain the rationality of the PBE1PBE method and LANL2DZ/6-31G(d) basis set, complex 2 was selected to do the calculation test with different functionals and basis sets. Tables 1 and 2 show that the excitation energies and ground geometries obtained by the basis set LANL2DZ/6-31G(d) are more accurate than the results obtained by other larger basis sets; moreover, LANL2DZ/6-31G(d) is good for saving computational resources. The geometry parameters and the excitation energies of absorptions obtained by different functionals including PBE1PBE, B3LYP (Becke’s three-parameter functional and the Lee-Yang-Parr functional) [38, 39], B3P86 (Becke’s three-parameter functional and the Perdew 86 functional), BPBE (Becke’s Perdew-Burke-Erzenrhof) [40], and BPW91 (Becke’s functional and the Perdew-Wang 91 functional) [41–43] were shown in Tables S1 and S2 (Supplementary material). The stable geometries and the absorptions obtained by PBE1PBE are more accurate than other functionals compared with the experimental results. Therefore, we predicted the structure and photophysical properties of complexes 1, 3 and 4 using the PBE1PBE method and LANL2DZ/6-31G(d) basis set.
Results and discussion
The molecular geometry structures in the ground state
The main optimized ground state geometry parameters of 1−5 together with the X-ray crystal diffraction data [19] of 2 are given in Table 3, and the optimized geometries of 1−5 in the gas phase are shown in Fig. 1.

Optimized ground state geometries structure of 1−5 at the PBE1PBE level
Table 3 indicates that the optimized bond lengths and bond angles of 2 are in general agreement with the experimental values. The geometry around Ir in each of the complexes is distorted octahedral with two mutually trans ppy pyridyl groups and two pyridine-derived monodentates, which are trans to the ppy phenyl rings. Due to the intense participation of a conjugating phenyl groups, the bond lengths of Ir − N3(4) are slightly longer than those of Ir − N1(2) [44]. All the Ir − C1(2) and Ir − N1(2) bond lengths and bond angles are within normal ranges in comparison with other Ir(III) bis-ppy complexes with two monodentate ligands [45–47]. The small change of Ir − N3(4) result from the effects of different ligands L. In addition, the strengthened metal − ligand (Ir −N) bond is significantly important to improve the phosphorescent quantum efficiency, and this may be the origin of the high efficiency of these complexes [48].
Electronegativity of the ligands L
In order to study the relationship between electron-withdrawing abilities of different ligands L (4-pyCN, 4-pyCHO, 4-pyCl, py, and 4-pyNH2) and electronic structure, the electronegativity (χ) was introduced to discuss. It can be expressed as [49]: χ = −(E HOMO + E LUMO)/2. Here, E HOMO is the energy of highest occupied molecular orbital (HOMO), and E LUMO is the energy of lowest unoccupied molecular orbital (LUMO). From the HOMO and LUMO energies of optimized ligands L, the calculated χ values are 4-pyCN (4.94 eV) > 4-pyCHO (4.85 eV) > 4-pyCl (4.21 eV) > py (3.82 eV) > 4-pyNH2 (3.10 eV) (Table S3 in Supplementary material), indicating that the electron-withdrawing abilities of the ligands L decrease along this order.
The frontier molecular orbital properties
The frontier molecular orbital compositions of 1−5 are compiled in Tables S4−S8 (Supplementary material). Schematic diagrams of the HOMOs and the LUMOs of 1−5 are shown in Fig. 2. The calculated results showed that LUMO and LUMO + 1 of 1 and 2 are localized on the π*(L). For 3, π*(ppy) and π*(L) contribute the compositions of LUMO and LUMO + 1 as shown in Table S6 (Supplementary material) and Fig. 2. However, the LUMO and LUMO+1 of 4 and 5 are mostly concentrated on π*(ppy). It is interesting to note that the compositions of LUMO and LUMO+1 convert from π*(L) to π*(ppy) with decreased χ values of L ligands 1 > 2 > 3 > 4 > 5. In contrast, the compositions of higher occupied molecular orbitals are hardly affected by L ligands. The HOMOs of 1−5 have strong d(Ir) (> 45 %) which is nearly equivalent contribution from the π(ppy). The HOMO−1 s of 1−5 lie primarily on the π(ppy) (> 85 %), with the exception that the HOMO−1 of 5 is composed of d(Ir), π(ppy), and π(L). Moreover, the contribution of π(ppy) to the HOMO-1 decreases in the order of 1 > 2 > 3 > 4 > 5, in line with the electron-withdrawing abilities of L ligands: 4-pyCN > 4-pyCHO > 4-pyCl > py > 4-pyNH2.

Schematic diagrams of the HOMOs and the LUMOs of 1−5 (Isovalue for orbital surface = 0.05)
Different L ligands can change the energy levels of the LUMO more significantly than those of the HOMO, and the LUMO energies increase with decreased χ values of ligands L: 1 (−2.9779 eV) < 2 (−2.8234 eV) < 3 (−1.8828 eV) < 4 (−1.7549 eV) < 5 (−1.6277 eV). Therefore, the strong electronegativity group L can stabilize the LUMO and make HOMO-LUMO energy gap narrow [50, 51]. On the contrary, the weak electronegativity group 4-pyNH2 increases the LUMO energy in 5. In addition, Table S9 (Supplementary material) shows that the solvent effect is obvious, and the energies of HOMO and LUMO of 1−5 are greatly decreased in gas phase. The effects of ligands L are different from those of ligands X in complexes [Ir(ppy)2X2] (X = CN, NCS, and NCO) [52], in which the energy levels of HOMO are changed more significantly than those of LUMO by tuning X ligands.
Absorptions in CH2Cl2 media
The calculated absorption energies associated with their oscillator strengths, the main configurations, and their assignments, as well as the experimental results of 2 and 5 are given in Table 4. The fitted Gaussian type absorption curves shown in Figs. 3 and 4 display the energy levels of molecular orbital involved in transitions of 1−5, which can intuitively understand the transition process.

Simulated absorption spectra of 1−5 in CH2Cl2 media with the calculated data at the TD-DFT level

Diagrams of the molecular orbital related to the absorptions of 1−5
In the visible region (420–520 nm), the absorption bands of 1 and 2 at 457 nm and 472 nm are contributed by HOMO → LUMO excitation. Table S5 (Supplementary material) shows that HOMO of 2 is composed of d(Ir) and π(ppy), while LUMO is dominantly localized on L (4-pyCHO) ligand. Thus, the absorption of 2 at 472 nm is attributed to {[d(Ir) + π(ppy)] → [π*(L)]} transition with metal-to-ligand and ligand-to-ligand charge transfer (MLCT/LLCT) transition characters. Similarly, the absorption of 1 at 457 nm is assigned to {[d(Ir) + π(ppy)] → [π*(L)]} (L = 4-pyCN) transition. For complexes 3−5, there are not strong absorptions in this region.
Figure 3 shows there are distinguishable bands at 320–420 nm for 1−5. Table 4 shows that the electron excitation from H − 2 → L (CI = 0.59458) and H − 4 → L (CI = 0.29439) should be responsible for the distinguishable singlet → singlet absorption band at 363 nm of 2. Table S5 shows that both H − 2 and H − 4 of 2 are composed of \( {{\text{d}}_{{{z^2}}}}\left( {\text{Ir}} \right) \), d xz (Ir), \( {d_{{{x^2} - {y^2}}}}\left( {\text{Ir}} \right) \), and π(ppy), whereas the LUMO is dominantly localized on π*(L). Thus, the absorption at 363 nm for 2 can be assigned to \( \left\{ {\left[ {{{\text{d}}_{{{z^2}}}},{{\text{d}}_{{xz}}}\left( {\text{Ir}} \right),{d_{{{x^2} - {y^2}}}}\left( {\text{Ir}} \right) + \pi \left( {\text{ppy}} \right)\left] \to \right[\pi *\left( {\text{L}} \right)} \right]} \right\} \) transition with mixing MLCT/LLCT (Fig. 4). Similarly, the absorption at 353 nm of 1 contributed by H − 2 → L (CI = 0.60453) and H − 4 → L (CI = 0.33736) can be described as MLCT/LLCT character, but the absorption bands of 3−5 have different transition nature. Table S6 (Supplementary material) and Fig. 4 show that the calculated absorption at 373 nm of 3 is produced by the two excitations of H → L + 1 and H → L + 2 with the configuration coefficients of 0.55231 and 0.40505, and can be characterized as \( \left\{ {\left[ {{{\text{d}}_{{{z^2}}}},{{\text{d}}_{{xz}}}\left( {\text{Ir}} \right),{ }{{\text{d}}_{{{x^2} - {y^2}}}}\left( {\text{Ir}} \right) + \pi \left( {\text{ppy}} \right)\left] \to \right[\pi *\left( {\text{ppy}} \right) + \pi *\left( {\text{L}} \right)} \right]} \right\} \) transition with MLCT/LLCT/intraligand charge transfer (ILCT) character. With respect to 4 and 5, the absorptions at 375 and 383 nm are contributed by electron excitation from {[d(Ir) + π(ppy)] → [π*(ppy)]} (HOMO → LUMO) with MLCT/ILCT character (Tables S7 and S8 in Supplementary material). It is found that the transition character of 1−5 converts from MLCT/LLCT to MLCT/ILCT with decreased χ values of L ligands. By comparing the absorptions of 1−5 at 353, 363, 373, 375, and 383 nm, it is found that the absorptions in this region are red-shifted in the order 1, 2, 3, 4, 5, which is consistent with a decreasing trend of χ value: 4-pyCN > 4-pyCHO > 4-pyCl > py > 4-pyNH2.
In the high energy region (220–320 nm), the shoulder absorptions of 1−5 can be assigned to {[d(Ir) + π(ppy)] → [π* (ppy) + π*(L)]} with MLCT/LLCT/ILCT transition character. Table 4 and Fig. 3 show that the highest energy absorptions of 2 at 218 nm is attributed to {π(ppy) + π(L)] → [π*(L)]} transition with LLCT/ILCT transition character. Similarly, the absorptions of 1 and 3 are dominantly assigned to LLCT/ILCT transition character mixed with MLCT component. For 4 and 5, {π(ppy) + π(L)] → [π*(ppy)]} excitation is in charge of the absorptions at 220 and 217 nm with MLCT/LLCT/ILCT transition character.
Experimentally, the absorptions of 2 and 5 at 350–500 nm are all assigned to MLCT transition, and the absorption of 5 is red-shifted compared with that of 2. In the next lower energy (280–350 nm), the shoulder absorptions can be ascribed to the π → π* and MLCT transitions, while in the ultraviolet region, measured at about 250 − 280 nm, the absorption bands are assigned to π → π* transitions. Furthermore, our calculated absorption bands of 2 and 5 are generally consistent with the measured excitation energy values and the transition assignments.
Geometry structures in the triplet excited state and emissions in CH2Cl2 media
The main geometry structural parameters of 1−5 in the lowest triplet states T1 optimized by the UPBE1PBE method are depicted in Table S10 (Supplementary material). The calculated results reveal that the structures of 1−5 do not vary notably relative to those of the ground states. The Ir − N(1) and Ir − N(2) bond lengths are relatively longer by about 0.007 Å and Ir − C bond lengths strengthen by about 0.030 Å. Due to the different electronegativity of ligands L, the Ir − N(3) and Ir − N(4) bond lengths in 1 and 2 are slightly shortened compared with those in the ground state, while those in 3−5 are somewhat elongated. The minor changes of bond lengths corresponds to the electrons being promoted from the Ir − ppy bonding orbital to the π* (ppy or L) orbital upon excitation.
The phosphorescence energies of 1–5 in CH2Cl2 media were calculated from the <DELTA>SCF method [53]: the energy difference between the ground singlet and triplet states in the triplet state optimized geometry. Compared with the experimental phosphorescence value of 2 and 5, it is more accurate than the excitation energies calculated by TDDFT (Table 5). It may be caused by the without spin-orbital coupling (SOC) effect in TD-DFT results. However, TDDFT can still provide a reasonable spectral feature for transition-metal complex systems [54, 55]. The frontier molecular orbital compositions responsible for the emissions are compiled in Table S11 (Supplementary material). As shown in Table 5, the lowest-energy emissions of 1−5 are mainly from the transitions of LUMO → HOMO. The analysis of the transition reveals that the emission of 2 at 553 nm originates from the 3{[d(Ir) + π(ppy)][π*(L)]} excited state with 3MLCT/3LLCT character. The nature of the phosphorescence of 1 is similar to that of 2, but the emissions of 3−5 have different transition nature. It is not L ligand but ppy ligand correlating with the emissions. With respect to 3, a 3MLCT/3ILCT/3LLCT type transition at 520 nm is described as 3{[d(Ir) + π(ppy)][π*(ppy) + π*(L)]}. The emissions of 4 and 5 have a combined 3MLCT/3ILCT character and should come from the 3{[d(Ir) + π(ppy)][π*(ppy)]} excited state. Herein, we note that the ligand L (4-pyCN or 4-pyCHO) with large electronegativity has a great effect on the phosphorescence character (3MLCT/3LLCT), but the small electronegativity ligand L (4-pyNH2) hardly changes the transition character compared with the emission character of 4 (3MLCT/3ILCT). In addition, the different phosphorescence character has brought about changes in quantum yield and lifetime, which of 5 have been improved or lengthened compared with that of 2 [19].
The above discussion reveals that the absorptions calculated at 353, 363, 373, 375, and 383 nm for 1−5, respectively, dominantly arise from the combination of MLCT and LLCT or ILCT electronic transitions, while the calculated phosphorescences are just the reverse process of the absorptions. Furthermore, we note that the compositions of HOMO are similar for 1−5, while those of LUMOs are different. The HOMOs of 1 and 2 are populated on L ligands but those of 4 and 5 are localized on ppy ligand. Therefore, the calculation results indicate that there is a competition between two different types of 3MLCT transitions: one type concentrates on the L ligand, and the other type relates to the ppy ligand, and which one will win the competition depends on the electronegativity of the ligand L. Hay [56] came to a similar conclusion that two types of 3MLCT involved a π* orbital on the C^N or LX ligands compete in the emission.
Solvent effect on the absorption and emission spectra
Different solvents may cause different excitation energies due to the polarity. However, very similar emission spectra of 2 were observed in different solvents (dichloromethane and acetone) within the experimental errors (Table 5). In order to discuss the solvent effect deeply, absorptions and emissions of 1−5 are evaluated with PCM method in methanol, acetone, chloroform, toluene, and cyclohexane, as shown in Table S12 (Supplementary material). It is obvious that both the absorption and emission spectra are very similar in different solvents. Hence, no solvent effects on the absorptions and emissions were observed, as found for some bis-cyclometallated complexes containing three-atom chelates [Ir(η2-ppy)2(XZY)] (XZY− = mp−, mhp−, chp−, ac−, bz−, ma−, tma−) [57]. We think the knowledge that the Iridium (III) complexes [Ir(C^N)2(L)2]+ have similar absorptions and emissions in different solvents will provide useful guidance for future experiments.
Conclusions
A series of cationic iridium (III) complexes [Ir(ppy)2(L)2]+ were investigated theoretically containing different pyridine derivatives L. The ligands L with different electronegativity can cause some variations in electronic structures and spectroscopic properties.
-
1.
The nature of LUMO and LUMO + 1 change from π*(L) to π*(ppy) with decreased electronegativity (χ) values of ligands L, but that of HOMO is hardly affected.
-
2.
The absorption transition character of 1−5 converts from MLCT/LLCT to MLCT/ILCT and the absorptions are red-shifted in the order 1, 2, 3, 4, 5, which is consistent with a decreasing trend of χ value: 4-pyCN > 4-pyCHO > 4-pyCl > py > 4-pyNH2.
-
3.
The different phosphorescence character (3MLCT/3LLCT or 3MLCT/3ILCT) has brought about changes in quantum yield and lifetime.
-
4.
The iridium (III) complexes [Ir(C^N)2(L)2]+ have similar absorptions and emissions in different solvents.
We hope that the study could provide useful information for the design of new phosphors in chemosensors with high phosphorescence quantum yields.
References
Tsuboyama A, Iwawaki H, Fururori M, Mukaide T, Kamatani J, Igawa S, Moriyama T, Miura S, Takiguchi T, Okada S, Hoshino M, Ueno K (2003) Homoleptic cyclometalated iridium complexes with highly efficient red phosphorescence and application to organic light-emitting diode. J Am Chem Soc 125:12971–12979
Adachi C, Baldo MA, Thompson ME, Forrest SR (2001) Nearly 100 % internal phosphorescence efficiency in an organic light emitting device. J Appl Phys 90:5048–5051
Lo KKW, Ng DCM, Chung CK (2001) First examples of luminescent cyclometalated iridium(III) complexes as labeling reagents for biological substrates. Organometallics 20:4999–5001
Gao R, Ho DG, Hernandez B, Selke M, Murphy D, Djurovich PL, Thompson ME (2002) Bis-cyclometalated Ir(III) complexes as efficient singlet oxygen sensitizers. J Am Chem Soc 124:14828–14829
Goldsmith JL, Hudson WR, Lowry MS, Anderson TH, Bernhard S (2005) Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. J Am Chem Soc 127:7502–7510
Kapturkiewez A, Angulo G (2003) Extremely efficient electrochemiluminescence systems based on tris(2-phenylpyridine)iridium(III). J Chem Soc Dalton Trans 3907–3913
Liu T, Zhang HX, Shu X, Xia BH (2007) Theoretical studies on structures and spectroscopic properties of a series of novel mixed-ligand Ir(III) complexes [Ir(Mebib)(ppy)X]. Dalton Trans. 1922–1928
Ikai M, Tokito S, Sakamoto Y, Suzuki T, Taga Y (2001) Highly efficient phosphorescence from organic light-emitting devices with an exciton-block layer. Appl Phys Lett 79:156–158
Liu T, Xia BH, Zhou X, Zhang HX, Pan QJ, Gao JS (2007) Theoretical studies on structures and spectroscopic properties of bis-cyclometalated iridium complexes. Organometallics 26:143–149
Ma DL, Wong WL, Chung WH, Chan FY, So PK, Lai YCL, Wong KY (2008) A highly selective luminescent switch-on probe for histidine/histidine-rich proteins and its application in protein staining. Angew Chem Int Ed 47:3735–3739
Carraway ER, Demas JN, DeGraff BA, Bacon JR (1991) Photophysics and photochemistry of oxygen sensors based on luminescent transition-metal complexes. Anal Chem 63:337–342
Liu T, Zhang HX, Zhou X, Zheng QC, Xia BH, Pan QJ (2008) Mechanism of Ir(ppy)2(N^N)+ (N^N = 2-Phenyl-1H-imidazo[4,5-f][1, 10]phenanthroline) sensor for F−, CF3COOH, and CH3COO−: density functional theory and time-dependent density functional theory studies. J Phys Chem A 112:8254–8262
Zhao Q, Li FY, Liu SJ, Yu MX, Liu ZQ, Yi T, Huang CH (2008) Highly selective phosphorescent chemosensor for fluoride based on an iridium(III) complex containing arylborane units. Inorg Chem 47:9256–9264
Goodall W, Williams JAG (2000) Iridium(III) bis-terpyridine complexes incorporating pendent N-methylpyridinium groups: luminescent sensors for chloride ions. J Chem Soc Dalton Trans 2893–2895
Amao Y, Ishikawa Y, Okura I (2001) Green luminescent iridium(III) complex immobilized in fluoropolymer film as optical oxygen-sensing material. Anal Chim Acta 445:177–182
Di Marco G, Lanza M, Pieruccini M, Campagna S (1996) A luminescent iridium(III) cyclometallated complex immobilized in a polymeric matrix as a solid-state oxygen sensor. Adv Mater 8:576–580
Zhao Q, Cao TY, Li FY, Li XH, Huang CH (2007) A highly selective and multisignaling optical-electrochemical sensor for Hg2+ based on a phosphorescent iridium(III) complex. Organometallic 26:2077–2081
Zhao N, Wu YH, Zhang X, Chen ZN (2009) Conversion from ILCT to LLCT/MLCT excited state by heavy metal ion binding in iridium(III) complexes with functionalized 2,2΄-bipyridyl ligands. Organometallics 28:5603–5611
Sie WS, Lee GH, Tsai KYD, Chang IJ, Shiu KB (2008) Synthesis, structures, properties, and chemical-sensor application of iridium(III) bis-cyclometallated complexes with two pyridine-derived ligands. J Mol Struct 890:198–202
Runge E, Gross EKU (1984) Density-functional theory for time-dependent systems. Phys Rev Lett 997–1000
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick KA, Rabuck D, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Hallacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C. 02. Gaussian Inc, Wallingford
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3865
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6170
Stratmann RE, Scuseria GE (1998) An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J Chem Phys 109:8218–8224
Matsuzawa NN, Ishitani A (2001) Time-dependent density functional theory calculations of photoabsorption spectra in the vacuum ultraviolet region. J Phys Chem A 105:4953–4962
Casida ME, Jamorski CKC, Casida DR (1998) Molecular excitation energies to high-lying bound states from timedependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J Chem Phys 108:4439–4449
Cossi M, Scalmani G, Regar N, Barone V (2002) New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J Chem Phys 117:43–54
Barone V, Cossi M (1997) A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J Chem Phys 107:3210–3221
Ros P, Schuit GCA (1966) molecular orbital calculations on copper chloride complexes. Theor Chim Acta (Berl) 4:1–12
Gorelsky SI (2010) SWizard program, revision 4.6. http://www.sg-chem.net/, University of Ottawa, Ottawa, Canada
Hay PJ, Wadt WR (1985) Ab Initio effective core potentials for molecular calculations. Potentials for the transition metal atoms. Sc to Hg J Chem Phys 82:270–283
Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculationsPotentials for K to Au including the outermost core orbitals. J Chem Phys 82:299–310
Liu T, Xia BH, Zheng QC, Zhou X, Pan QJ, Zhang HX (2010) DFT/TD-DFT investigation on Ir(III) complexes with N-Heterocyclic carbene ligands: geometries, electronic structures, absorption, and phosphorescence properties. J Comput Chem 31:628–638
Zhao Q, Liu SJ, Shi M, Li FY, Jing H, Yi H, Huang CH (2007) Tuning photophysical and electrochemical properties of cationic iridium(III) complex salts with imidazolyl substituents by proton and anions. Organometallics 26:5922–5930
Di Censo D, Fantacci S, De Angelis F, Klein C, Evans N, Kalyanasundaram K, Bolink HJ, Gratzel M, Nazeeruddin MK (2008) Synthesis, characterization, and DFT/TD-DFT calculations of highly phosphorescent blue light-emitting anionic iridium complexes. Inorg Chem 47:980–989
Zhao Q, Liu SJ, Shi M, Wang CM, Yu MX, Li L, Li FY, Yi T, Huang CH (2006) Series of new cationic iridium(III) complexes with tunable emission wavelength and excited state properties: structures, theoretical calculations, and photophysical and electrochemical properties. Inorg Chem 45:6152–6160
Bai F-Q, Zhou X, Liu T, Zhao G-J, Zhang J-P, Zhang H-X (2008) Computational studies on the spectroscopic properties of the 2-pyridylpyrazolate-based platinum(II) complexes with modified pyrazolate fragment. Int J Quantum Chem 109:308–319
Becke AD (1993) Densityfunctional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B 45:13244–13249
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B 46:6671–6687
Levy M, Perdew JP (1993) Tight bound and convexity constraint on the exchange-correlation-energy functional in the low-density limit, and other formal tests of generalized-gradient approximations. Phys Rev B 48:11638–11645
Coe BJ, Glenwright SJ (2000) Trans-effects in octahedral transition metal complexes. Coord Chem Rev 203:5–80
DeRosa MC, Mosher PJ, Yap GPA, Focsaneanu K-S, Crutchley RJ, Evans CEB (2003) Synthesis, characterization, and evaluation of [Ir(ppy)2(vpy)Cl] as a polymer-bound oxygen sensor. Inorg Chem 42:4864–4872
Chin CS, Eum M-S, Kim SY, Kim C, Kang SK (2006) New type of photoluminescent iridium complex: novel synthetic route for cationic trans-bis(2-phenylpyridinato)iridium(III) complex. Eur J Inorg Chem 24:4979–4982
Chin CS, Eum M-S, Kim SY, Kim C, Kang SK (2007) Blue-light-emitting complexes: cationic (2-phenylpyridinato)iridium(III) complexes with strong-field ancillary ligands. Eur J Inorg Chem 3:372–375
Li XN, Wu ZJ, Liu XJ, Zhang HJ (2010) Origin of rare and highly efficient phosphorescent and electroluminescent iridium(III) complexes based on C^N = N ligands, a theoretical explanation. J Phys Chem A 114:9300–9308
Zhan CG, Nichols JA, Dixon DA (2003) Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J Phys Chem A 107:4184–4195
Cherry WR, Henderson LJ (1984) Relaxation processes of electronically excited states in polypyridine ruthenium complexes. Inorg Chem 23:983–986
Lumpkin RS, Kober EM, Worl LA, Murtaza Z, Meyer TJ (1990) Metal-to-ligand charge-transfer (MLCT) photochemistry: experimental evidence for the participation of a higher lying MLCT state in polypyridyl complexes of ruthenium(II) and osmium(II). J Phys Chem 94:239–243
Liu T, Zhang H-X, Xia B-H (2008) Theoretical studies on structures and spectroscopic properties of bis-cyclometalated iridium complexes [Ir(ppy)2X2]−. J Organomet Chem 693:947–956
Guillon T, Boggio-Pasqua M, Alary F, Heully J-L, Lebon E, Sutra P, Lgau A (2010) Inorg Chem 49:8862–8872
Li XN, Liu XJ, Wu ZJ, Zhang HJ (2008) DFT/TDDFT studies on the electronic structures and spectral properties of rhenium(I) pyridinybenzoimidazole complexes. J Phys Chem A 112:11190–11197
Shi LL, Liao Y, Zhao L, Su ZM, Kan YH, Yang GC, Yang SY (2007) Theoretical studies on the electronic structure and spectral properties of versatile diarylethene-containing 1,10-phenanthroline ligands and their rhenium(I) complexes. J Organomet Chem 692:5368–5374
Hay PJ (2002) Theoretical studies of the ground and excited electronic states in cyclometalated phenylpyridine Ir(III) complexes using density functional theory. J Phys Chem A 106:1634–1641
Sie WS, Jian JY, Su TC, Lee GH, Lee HM, Shiu KB (2008) Synthesis, structures, and properties of iridium(III) bis-cyclometallated complexes containing three-atom chelates. J Organomet Chem 693:1510–1517
Acknowledgments
The authors are grateful to the financial aid from the National Nature Science Foundation of China (21031003) and Shanxi Natural Science Foundation (2010011012-2).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Tables giving additional optimized geometry parameters, orbital compositions and TD-DFT calculations.
ESM 1
(DOC 416 kb)
Rights and permissions
About this article
Cite this article
Zhang, TT., Qi, XX., Jia, J. et al. Tuning electronic structure and photophysical properties of [Ir(ppy)2(py)2]+ by substituents binding in pyridyl ligand: a computational study. J Mol Model 18, 4615–4624 (2012). https://doi.org/10.1007/s00894-012-1462-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1462-8