
Abstract
By using the density functional theory (DFT) and time-dependent density functional theory (TDDFT), the electronic structure and photophysical properties of a series of cyclometalated iridium(III) complexes bearing the substituted phenylpyrazole have been theoretically investigated. All studied iridium(III) complexes have the distorted octahedral geometry with cis-C,C, cis-O,O, and trans-N,N chelate disposition. The lowest lying singlet → singlet absorptions of all studied iridium(III) complexes are respectively located at 405 nm, 387 nm, 382 nm, 370 nm, and 387 nm. The calculated emission wavelengths for all studied iridium(III) complexes are 654 nm, 513 nm, 506 nm, 505 nm and 499 nm, respectively. The calculated emission wavelength for complex 4 at the CAM-B3LYP level is in good agreement with the experimental value. From the theoretical results, it can be seen that the electron-donating substituent groups have the important effect on the electronic structure and photophysical properties of all studied iridium(III) complexes. We hope that this study can provide valuable guidance for the design of new phosphorescent organic light-emitting diodes (OLEDs) materials.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the last two decades, due to the benefits of relatively short excited-state lifetimes, high photoluminescence efficiency, good colour tunability, general thermal, and electrochemical stability, phosphorescent transition metal complexes of Ir(III), Pt(II), Os(II), Cu(I) and so on have been widely explored [1,2,3,4,5,6]. Especially, phosphorescent cyclometallated iridium(III) complexes have already been extensively investigated for many applications, such as organic light-emitting diodes (OLEDs), solar cells, sensors, and bioimaging [7,8,9,10,11,12]. The phosphorescence quantum yield is enhanced beyond that achievable with fluorescent emitters due to the strong spin–orbit coupling induced by the iridium atom, which allows for mixing of singlet and triplet excited states through efficient intersystem crossing [13, 14].
Many researchers have extensively investigated the color tuning via the molecular design of Ir(III) complexes to realize full-color displays. Generally speaking, one strategy is to vary the heterocycle, the degree of conjugation in the C^N ligand, and/or the ancillary ligand. Another strategy is to alter substituents on the cyclometalated phenyl, the coordination heterocycle, or the ancillary ligands. F.Q. Bai et al. have designed and studied many iridium(III) complexes with different substituent groups, which indicates that the substituent groups could be propitious to produce better performance of iridium(III) materials [15, 16]. B.Q. Liu et al. have studied a series of neutral cyclometalated Ir(III) complexes bearing substituted N‑heterocyclic carbene (NHC) ligands for high-performance yellow OLED application [17]. The influence of the electron-donating or electron-withdrawing substituents on their photophysical properties have been explored. In recent years, tetraphenylimidodiphosphinate (tpip) derivatives with polar P=O bonds and phenyl rings have been used as ancillary ligands to obtain Ir(III) complexes for efficient OLEDs because they could increase the electron mobility of the complexes and improve their corresponding electroluminescent performances [18, 19]. L.M. Groves et al. have studied a series of Ir(III) complexes based on phenylpyrazole (ppz) derivatives with electron-withdrawing groups (–F, –CF3, –OCF3, and –SF5), which revealed a broad, featureless emission peak that ranged between 510 and 560 nm [20]. Z.G. Niu et al. have synthesized and fully characterized seven cyclometalated iridium(III) complexes Ir1–Ir7 based on phenylpyrazole derivatives as main ligands and tetraphenylimidodiphosphinate (tpip) as the ancillary ligand [21]. Via variation of the main ligands, the emission colors for complexes Ir1–Ir7 can be adjusted from blue to yellow in CH2Cl2 solutions. It is known that the control of the optical physical process must ensure the structural similarity, so that the expected increase of the radiative decay rate can ensure the improvement of the overall efficiency [22, 23]. Hence, on the basis of complex Ir6 [21], we have theoretically designed four complexes and investigated their electronic structure and photophysical properties by density functional theory (DFT) and time-dependent density functional theory (TDDFT) method.
2 Computational method
The ground state geometry for each molecule was optimized by density functional theory (DFT) method with hybrid Hartree–Fock/density functional model (PBE0) based on the Perdew–Burke–Erzenrhof (PBE) [24, 25]. On the basis of the ground- and excited-state equilibrium geometries, the time-dependent DFT (TDDFT) approach was applied to investigate the absorption and emission spectral properties. The “double-ξ” quality basis set LANL2DZ associated with the pseudopotential was employed on atom Ir [26, 27]. The 6–31G(d,p) basis set was used for nonmetal atoms in the gradient optimizations. Furthermore, the stable configurations of these complexes can be confirmed by frequency analysis, in which no imaginary frequency was found for all configurations at the energy minima. All calculations were performed with the polarized continuum model (PCM) in CH2Cl2 medium. All calculations were performed with the Gaussian 09 software package [28].
3 Results and discussion
3.1 Geometries in the ground state S0 and triplet excited state T1
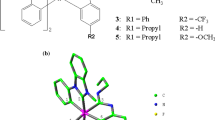
The sketch map of iridium(III) complexes 1–5 have been presented in Fig. 1a, and the optimized ground state geometric structure for complex 1 is shown in Fig. 1b along with the numbering of some key atoms. The main optimized geometric parameters of the ground state (S0) and the lowest lying triplet excited state (T1) have been summarized in Table 1.

a Sketch map of the structures of iridium(III) complexes 1–5. b Vertical view (left) and side view (right) of the optimized geometrical structure of iridium(III) complex 1
These cyclometalated iridium(III) complexes are based on the substituted phenylpyrazole derivatives as main ligands and tetraphenylimidodiphosphinate (tpip) as the ancillary ligand. The coordination geometry of the iridium center in each complex is a distorted octahedral geometry with cis-C,C, cis-O,O, and trans-N,N chelate disposition. The lengths of Ir–C1, Ir–C2, Ir–N1 and Ir–N2 of complex 4 are close to those in reported data [21]. The lengths of Ir–C1 and Ir–C2 of complexes 1–5 are 1.98 or 1.99 Å. The lengths of Ir–N1 and Ir–N2 of complexes 1–5 are 2.00 or 2.01 Å. The lengths of Ir–O1 and Ir–O2 of complexes 1–5 are 2.23 or 2.24 Å. The bond angles C1–Ir–O2, C2–Ir–O1 and N1–Ir–N2 of complexes 1–5 are at the range from 170° to 177°. The dihedral angles C1–N1–O2–N2 and C2–N1–O1–N2 of complexes 1–5 are smaller than 3°, which indicates that all these iridium complexes possess a distorted octahedral configuration.
Beside, to investigate the changes of the geometrical structures upon excitation, the calculated geometrical parameters of T1 state for complexes 1–5 have been provided in Table 1. The bond lengths Ir–C1 and Ir–C2 of complexes 1–5 in T1 state are slightly smaller than those in S0 state. The bond angles C1–Ir–O2, C2–Ir–O1 and N1–Ir–N2 of complexes 1–5 in T1 state have slight change in contrast to those in S0 state. The dihedral angle C1–N1–O2–N2 of complexes 1–5 in T1 state are larger than those in S0 state. It is interesting to note that the dihedral angle C2–N1–O1–N2 of complex 1 in T1 state is smaller than that of S0 state. However, the dihedral angles C2–N1–O1–N2 of complex 2–5 in T1 state are obviously larger than those of S0 state.
3.2 Molecular orbital properties
The frontier molecular orbital (FMO) components and the energy level have been investigated to describe the electronic and photophysical properties. Figure 2 shows the HOMOs and LUMOs distribution (iso-surface value 0.02), energy levels, and energy gaps between of LUMO and HOMO (ΔEL→H) of the complexes 1–5. The detailed information of FMO compositions for complexes 1–5 at the PBE0/6–31G(d,p) + LANL2DZ level has been listed in Tables S1–S5 (Supplementary Materials). It can be seen from Fig. 2 and Tables S1–S5 (Supplementary Materials) that the HOMO of complex 1 resides mainly on the L1 ligand, however, the HOMO of complexes 2–5 are mainly located at Ir atom and L1 ligand. For example, the HOMO of 2 distributes over the d-orbital of Ir (34%) and the π-orbital of L1 ligand (63%). The LUMO of complexes 1–5 resides mostly on the L1 ligand, for example, complex 1 has the 93% antibonding π*-orbital of the L1 ligand. It is known that the different R substituent groups on the main ligand have different electron-donating and electron-accepting abilities, that is, the order of electron-donating ability –N(CH3)2 > –CH3 > –H, the order of electron-accepting ability –H < –F < –NO2. In comparison with complex 1, it can be seen that the introduction of the electron-donating substituents (–N(CH3)2 and –CH3) or electron-accepting ones (–F and –NO2) can slightly raise or lower the HOMO and LUMO energy levels. It is particularly interesting to note that the order of HOMO or LUMO energy levels is 1 > 2 > 3 > 4 > 5. Obviously, the HOMO and LUMO energy levels of complex 1 are the largest ones among these studied complexes. Furthermore, the ΔEL→H value (3.78 eV) of 1 is the smallest one among complexes 1–5. In addition, complex 4 has the largest ΔEL→H value 4.22 eV in these studied complexes. Complex 5 has the smallest HOMO and LUMO level, that is, − 5.59 eV and − 1.67 eV, respectively. The HOMO of 5 distributes over the d-orbital of Ir (35%) and the π-orbital of L1 ligand (60%). Meanwhile, the LUMO of 5 resides mostly on the L1 ligand (99%).

Molecular orbital diagrams and HOMO and LUMO energies for complexes 1–5
3.3 Absorption spectra
The PBE0 method has been used to calculate the absorption properties of complexes 1–5 on the basis of the optimized ground state geometries. Table S6 (Supplementary Materials) presents the vertical electronic excitation energies, oscillator strengths (f), dominant orbital excitations and their assignments of the S0 and T1 states. The stimulated absorption spectra of complexes 1–5 in CH2Cl2 solvent based on the TDDFT calculations has been described in Fig. 3.

Simulated absorption spectra in CH2Cl2 medium for complexes 1–5
As shown in Table S6, the lowest lying singlet → singlet absorption of 1–5 are located at 405 nm (f = 0.1175), 387 nm (f = 0.0507), 382 nm (f = 0.0523), 370 nm (f = 0.0610) and 387 nm (f = 0.0208), respectively. It can be seen that the calculated absorption wavelength for complex 4 is comparable with the experimental value [19]. In contrast to complexes 2–5, complex 1 has an obviously large absorption peak at about 288 nm wavelength, which originates from the contributions HOMO → LUMO + 2 (34%) and HOMO → LUMO + 6 (25%) electron transition configurations. In addition, complex 1 has also a small absorption peak at 398 nm. The lowest lying absorption of complex 1 is obviously redshifted in contrast to that of complex 3, which is due to the strong electron-donating substituent group –N(CH3)2. Complexes 2–4 have the similar absorption curve shape with two peaks at about 243 nm and 310 nm, respectively. In contrast to that of complex 3, the lowest lying absorption of 1 is obviously red-shifted, which is consistent with the variation of the ΔEL→H values, which indicates that the substitute groups –N(CH3)2 has the obvious effect on the lowest lying singlet → singlet absorption wavelengths. The lowest lying absorption for complexes 1–5 have the different transition configuration contributing to the S0 → S1 state. For example, the lowest lying absorption for complex 1 has the HOMO → LUMO (39%) and HOMO → LUMO + 1 (49%) transition configuration and intraligand charge transfer (ILCT) [π(L1) → π*(L1)] character. The lowest lying absorption for complex 3 has the HOMO → LUMO + 1 (94%) transition configuration and metal to ligand charge transfer (MLCT)/ILCT character. This shows that the different substituent groups have important influence on the transition character. Complex 5 with the strong electron-accepting substituent –NO2 has the weakest absorption intensity in comparison with other four complexes. It can be seen that there is an obvious difference between the absorption spectrum of complex 1 and that of 5, which indicates that the electron-donating substituent –N(CH3)2 and electron-accepting substituent –NO2 have the different effect on the absorption properties of iridium(III) complexes.
3.4 Phosphorescence property
To check the computational method, five different density functionals (B3LYP [29], CAM-B3LYP [30], M052X [31], M062X [32], and PBE0 [33]) were used to calculate the emission of complex 4. A better agreement with experimental data was obtained for CAM-B3LYP relative to the other four functionals. The calculated emission wavelengths for complex 4 at B3LYP, CAM-B3LYP, M052X, M062X and PBE0 levels are 526, 505, 457, 436 and 526 nm, respectively, with the deviations of 32, 11, 37, 58 and 32 nm from the measured value of 494 nm [19]. It is obvious that the CAM-B3LYP functional leads to a more satisfactory result. Hence, we have employed the CAM-B3LYP functional for further emission spectral calculations. On the basis of the optimized triplet excited-state geometries, the emission properties of complexes 1–5 in CH2Cl2 medium obtained using the TDDFT/CAM-B3LYP method are shown in Table 2. For comparison, the available experimental value has also been listed in Table 2. The plots of the molecular orbitals related to emissions of complexes 1–5 have been depicted in Table 3. In addition, partial frontier molecular orbital compositions (%) of complexes 1–5 in the T1 state have been provided in Table S7 (Supplementary Materials).
From Table 2 it can be seen that the calculated lowest energy emissions of complex 1 is obviously red-shifted in comparison with that of 3, which shows electron-donating substituent group –N(CH3)2 has great effect on the phosphorescence emission wavelength. The emission transition characters for complex 1 are assigned to triplet intraligand charge transfer (3ILCT) [π*(L1) → π(L1)]. In addition, compared to complex 3, the calculated lowest energy emissions of 2 is also slightly red-shifted, which originates from the L → H (43%) and L + 1 → H–1 (24%) transition configuration with triplet metal to ligand charge transfer (3MLCT)/ triplet intraligand charge transfer (3ILCT) and 3ILCT characters. However, the introduction of electron-accepting substituent group –F in complex 4 has no obvious influence on the phosphorescence emission properties, that is, 3 and 4 have the similar phosphorescence emission properties. Besides, the introduction of electron-accepting substituent group –NO2 in complex 5 leads to the sight blueshift of phosphorescence emission wavelength, however, possessing the similar transition character to complex 3. As shown in Table 3 and Table S7, the HOMO and LUMO of complex 1 mainly distribute on the L1 ligand. However, the emission transition characters for complexes 2–5 are assigned to LUMO → HOMO and LUMO + 1 → HOMO–1 transition configurations characterized as triplet metal-to-ligand charge transfer (3MLCT) and 3ILCT. The HOMO of complexes 2–5 are mainly located on the Ir atom and L1 ligand. For example, the HOMO of complex 2 is distributed on the Ir atom (30%) and L1 ligand (66%). Therefore, the introduction of the different electron-donating substituents or electron-accepting ones probably have different effect on the phosphorescence properties.
3.5 Phosphorescence quantum yield
The emission quantum yield (Φ) can be affected by the competition between kr (radiative decay rate) and knr (nonradiative decay rate), i.e. Φ = kr/(kr + knr). To increase the quantum yield, kr should be increased and knr should be decreased simultaneously or respectively [34, 35]. Moreover, kr is also theoretically related to the mixing between S1 and T1, which is proportional to the spin–orbit coupling (SOC) and inversely proportional to the energy gaps between the S1 and T1 states according to the following formula [36, 37]:
where \(H_{{S_{0} }}\) is the Hamiltonian for the spin–orbit coupling, \(\mu_{{S_{1}^{{}} }}\) is the transition dipole moment in the S0 → S1 transition, \(\Delta E_{{{\text{S}}_{1} - {\text{T}}_{1} }}\) is the energy gaps between the S1 and T1 states, Eem represents the emission energy in cm−1 and n, h, and ε0 are the refractive index, Planck’s constant and the permittivity in a vacuum, respectively. Accordingly, the variation of quantum yield (Φ) can be qualitatively analyzed in theory from the above formula.
It is known that the phosphorescence quantum yield could be increased by a larger 3MLCT composition and thus the intersystem crossing (ISC). Additionally, for iridium atom, the direct involvement of the d(Ir) orbital enhances the first-order SOC in the T1 → S0 transition and thus ISC, which would result in a drastic decrease of the radiative lifetime and avoid the nonradiative process [38]. In Table 4, we have listed the 3MLCT contributions which were calculated to be 1.42%, 11.71%, 11.82%, 10.47% and 11.42% for complexes 1–5, respectively. The 3MLCT contribution of 1 is the smallest one among these complexes. It is also known that the phosphorescence quantum yield are inversely proportional to the \(\Delta E_{{{\text{S}}_{{1}} {\text{ - T}}_{{1}} }}\) [39]. Namely, a minimal \(\Delta E_{{{\text{S}}_{{1}} {\text{ - T}}_{{1}} }}\) is required for enhancing the ISC rate, leading to the increased kr. The \(\Delta E_{{{\text{S}}_{{1}} {\text{ - T}}_{{1}} }}\) for these complexes are also listed in Table 4, along with the \(\mu_{{S_{1}^{{}} }}\) values. The results give 0.6805, 1.4502, 0.4650, 0.5438 and 0.4762 eV for the \(\Delta E_{{{\text{S}}_{{1}} {\text{ - T}}_{{1}} }}\), respectively, for complexes 1–5. From the discussion above, it is obvious that a lower \(\Delta E_{{{\text{S}}_{{1}} {\text{ - T}}_{{1}} }}\) and larger 3MLCT contributions and higher \(\mu_{{S_{1}^{{}} }}\) values may account for a larger kr according to Eq. (1). F.Q. Bai et al. have concluded that electron-withdrawing substituent groups on cyclometalating ligands cannot completely decide the final quantum efficiency [23], which is also seen from complexes 4 and 5 in our study. From the data in Table 4, it can be seen that the complex 3 probably possesses the largest kr value among these studied complexes.
4 Conclusions
A series of cyclometalated iridium(III) complexes bearing the substituted phenylpyrazole have been studied by using DFT/TDDFT method to explore the electronic structure and photophysical properties. Theoretical calculation results show that the obtain bond lengths Ir–C1, Ir–C2, Ir–N1 and Ir–N2 of complex 4 from theoretical calculation have good agreement with the crystallographic data. The ΔEL→H values increase in the order of 1 (3.78 eV) < 5 (3.92 eV) < 2 (4.09 eV) < 3 (4.13 eV) < 4 (4.22 eV). Both the lowest lying singlet → singlet absorption wavelength and the lowest energy emission of complex 1 with the substitute groups –N(CH3)2 have the obvious redshift in contrast to other four complexes. According to the theoretical result, complex 3 could possesses the largest kr value among these studied complexes. It is anticipated that this study will be useful for designing novel Ir(III) complexes applied to OLEDs field.
References
Ding, Y., Liu, D., Li, J. Y., Li, H. T., Ma, H. Y., Li, D. L., & Niu, R. (2020). Saturated red phosphorescent Iridium(III) complexes containing phenylquinoline ligands for efficient organic light-emitting diodes. Dyes Pigments, 179, 108405.
Hsu, L. Y., Liang, Q. M., Wang, Z. H., Kuo, H. H., Tai, W. S., Su, S. J., Zhou, X. W., Yuan, Y., & Chi, Y. (2019). Bis-tridentate Ir-III phosphors bearing two fused five-six-membered metallacycles: A strategy to improved photostability of blue emitters. Chemistry-A European Journal, 25, 15375–15386.
Dragonetti, C., Fagnani, F., Marinotto, D., di Biase, A., Roberto, D., Cocchi, M., Fantacci, S., & Colombo, A. (2020). First member of an appealing class of cyclometalated 1,3-di-(2-pyridyl)benzene platinum(ii) complexes for solution-processable OLEDs. Journal of Materials Chemistry C, 8, 7873–7881.
Cheng, G., Kwak, Y., To, W. P., Lam, T. L., Tong, G. S. M., Sit, M. K., Gong, S. L., Choi, B., Choi, W. I., Yang, C. L., & Che, C. M. (2019). High-efficiency solution-processed organic light-emitting diodes with tetradentate platinum(II) emitters. ACS Journal of Materials Chemistry C, 11, 45161–45170.
Liao, J. L., Chi, Y., Yeh, C. C., Kao, H. C., Chang, C. H., Fox, M. A., Low, P. J., & Lee, G. H. (2015). Near infrared-emitting tris-bidentate Os(II) phosphors: Control of excited state characteristics and fabrication of OLEDs. Journal of Materials Chemistry C, 3, 4910–4920.
Song, Y. L., Jiao, B. J., Liu, C. M., Peng, X. L., Wang, M. M., Yang, Y., Zhang, B., & Du, C. X. (2020). Synthesis, structures and luminescent properties of red emissive neutral copper(I) complexes with bisphosphino-substituted benzimidazole. Inorganic Chemistry Communications, 112, 107689.
Cheng, W., Wang, L. D., Zhou, Y. Y., Bian, Z. Q., Tong, B. H., Liu, Z. W., & Wang, S. (2020). Blue iridium(III) complexes with high internal quantum efficiency based on 4-(pyridin-3-yl)pyrimidine derivative and their electroluminescent properties. Dyes Pigments, 177, 108257.
Liu, J., Ma, D. G., & Bai, J. (2019). Synthesis of a new iridium complex and its yellow efficient electroluminescence with low efficiency roll-off by exhaustive optimization of device. Journal of Luminescence, 215, 116655.
Pal, A. K., Krotkus, S., Fontani, M., Mackenzie, C. F. R., Cordes, D. B., Slawin, A. M. Z., Samuel, I. D. W., & Zysman-Colman, E. (2018). High-efficiency deep-blue-emitting organic light-emitting diodes based on iridium(III) carbene complexes. Advanced Materials, 30, 1804231.
Chen, Z., Wang, L. Q., Su, S. K., Zheng, X. Y., Zhu, N. Y., Ho, C. L., Chen, S. M., & Wong, W. Y. (2017). Cyclometalated iridium(III) carbene phosphors for highly efficient blue organic light-emitting diodes. ACS Applied Materials & Interfaces, 9, 40497–40502.
Sarada, G., Sim, B., Moon, C. K., Cho, W., Kim, K. H., Sree, V. G., Park, E., Kim, J. J., & Jin, S. H. (2016). Synthesis and characterization of highly efficient blue Ir(III) complexes by tailoring beta-diketonate ancillary ligand for highly efficient PhOLED applications. Organic Electronics, 39, 91–99.
Hwang, J., Yook, K. S., Lee, J. Y., & Kim, Y. H. (2015). Synthesis and characterization of phenylpyridine derivative containing an imide functional group on an iridium(III) complex for solution-processable orange-phosphorescent organic light-emitting diodes. Dyes Pigments, 121, 73–78.
Baldo, M. A., Lamansky, S., Burrows, P. E., Thompson, M. E., & Forrest, S. R. (1999). Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Applied Physics Letters, 75, 4–6.
Cleave, V., Yahioglu, G., Le Barny, P., Friend, R. H., & Tessler, N. (1999). Harvesting singlet and triplet energy in polymer LEDs. Advanced Materials, 11, 285–288.
Xie, L. M., Bai, F. Q., Li, W., Zhang, Z. X., & Zhang, H. X. (2015). Theoretical research on the effect of regulated π-conjugation on the photophysical properties of Ir(III) complexes. Physical Chemistry Chemical Physics: PCCP, 17, 10014–10021.
Wang, Y., Bai, F. Q., Ma, X. Y., & Zhang, H. X. (2018). A complete evaluation from theoretical aspect on the phosphorescent efficiency improvement through ancillary ligands modifications of a blue Ir(III) complex. Organic Electronics, 59, 293–300.
Liu, B. Q., Jabed, M. A., Guo, J. L., Xu, W., Brown, S. L., Ugrinov, A., Hobbie, E. K., Kilina, S., Qin, A. J., & Sun, W. F. (2019). Neutral Cyclometalated iridium(III) complexes bearing substituted N-heterocyclic carbene (NHC) ligands for high-performance yellow OLED application. Inorganic Chemistry, 58, 14377–14388.
Zhou, Y. H., Xu, Q. L., Han, H. B., Zhao, Y., Zheng, Y., Zhou, L., Zuo, J. L., & Zhang, H. J. (2016). Highly efficient organic light-emitting diodes with low efficiency roll-off based on iridium complexes containing pinene sterically hindered spacer. Advanced Optical Materials, 4, 1726–1731.
Zhou, Y. H., Xu, J., Wu, Z. G., & Zheng, Y. X. (2017). Synthesis, photoluminescence and electroluminescence of one iridium complex with 2-(2,4-difluorophenyl)-4-(trifluoromethyl) pyrimidine and tetraphenylimidodiphosphinate ligands. Journal of Organometallic Chemistry, 848, 226–231.
Groves, L. M., Schotten, C., Beames, J., Platts, J. A., Coles, S. J., Horton, P. N., Browne, D. L., & Pope, S. J. A. (2017). From ligand to phosphor: Rapid, machine-assisted synthesis of substituted iridium(III) pyrazolate complexes with tuneable luminescence. Chemistry—A European Journal, 23, 9407–9418.
Niu, Z. G., Han, H. B., Li, M., Zhao, Z., Chen, G. Y., Zheng, Y. X., Li, G. N., & Zuo, J. L. (2018). Tunable emission color of iridium(III) complexes with phenylpyrazole derivatives as the main ligands for organic light emitting diodes. Organometallics, 37, 3154–3164.
Wang, Y., Bao, P., Wang, J., Jia, R., Bai, F. Q., & Zhang, H. X. (2018). Comprehensive investigation into luminescent properties of Ir(III) complexes: An integrated computational study of radiative and nonradiative decay processes. Inorganic Chemistry, 57, 6561–6570.
Wang, Y., Wang, J., Zhang, H. X., Szilagyi, I. M., & Bai, F. Q. (2018). Strategies on cyclometalating ligand substitution of several Ir(III) complexes: Theoretical investigation of different molecular behaviors. Organometallics, 37, 2491–2499.
Hohenberg, P., & Kohn, W. (1964). Inhomogeneous electron gas. Physical Review, 136, B864–B871.
Adamo, C., & Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: The PBE0 model. The Journal of Chemical Physics, 110, 6158–6169.
Hay, P. J., & Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. The Journal of Chemical Physics, 82, 270–284.
Hay, P. J., & Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. The Journal of Chemical Physics, 82, 299–310.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., … Fox, D. J. (2009). Gaussian 09. Gaussian Inc.
Niehaus, T. A., Hofbeck, T., & Yersin, H. (2015). Charge-transfer excited states in phosphorescent organo-transition metal compounds: A difficult case for time dependent density functional theory? RSC Advances, 5, 63318–63329.
Cramer, C., & Truhlar, D. (1996). Solvent effects and chemical reactivity. Kluwer.
Zhao, Y., Schultz, N. E., & Truhlar, D. G. (2006). Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. Journal of Chemical Theory and Computation, 2, 364–382.
Zhao, Y., & Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06 functionals and 12 other functionals. Theoretical Chemistry Accounts, 120, 215–241.
Perdew, J. P., Burke, K., & Ernzerhof, M. (1996). Generalized gradient approximation made simple. Physical Review Letters, 77, 3865–3868.
Fantacci, S., De Angelis, F., Sgamellotti, A., Marrone, A., & Re, N. (2005). Photophysical properties of [Ru(phen)(2)(dppz)] (2+) intercalated into DNA: An integrated Car-Parrinello and TDDFT study. Journal of the American Chemical Society, 127, 14144–14145.
Tamayo, A. B., Garon, S., Sajoto, T., Djurovich, P. I., Tsyba, I. M., Bau, R., & Thompson, M. E. (2005). Cationic bis-cyclometalated iridium(III) diimine complexes and their use in efficient blue, green, and red electroluminescent devices. Inorganic Chemistry, 44, 8723–8732.
Haneder, S., Da Como, E., Feldmann, J., Lupton, J. M., Lennartz, C., Erk, P., Fuchs, E., Molt, O., Munster, I., Schildknecht, C., & Wagenblast, G. (2008). Controlling the radiative rate of deep-blue electrophosphorescent organometallic complexes by singlet-triplet gap engineering. Advanced Materials, 20, 3325–3330.
Turro, N. (1991). Modern molecular photochemistry. University Science Books.
Yang, C. H., Cheng, Y. M., Chi, Y., Hsu, C. J., Fang, F. C., Wong, K. T., Chou, P. T., Chang, C. H., Tsai, M. H., & Wu, C. C. (2007). Blue-emitting heteroleptic iridium(III) complexes suitable for high-efficiency phosphorescent OLEDs. Angewandte Chemie International Edition, 46, 2418–2421.
Avilov, I., Minoofar, P., Cornil, J., & De Cola, L. (2007). Influence of substituents on the energy and nature of the lowest excited states of heteroleptic phosphorescent Ir(III) complexes: A joint theoretical and experimental study. Journal of the American Chemical Society, 129, 8247–8258.
Acknowledgements
The authors are grateful to the financial aid from the Program of Science and Technology Development Plan of Jilin Province of China (Grant No. 20200201099JC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Han, D., Ji, X., Zhao, L. et al. Theoretical insight on electronic structure and photophysical properties of a series of cyclometalated iridium(III) complexes bearing the substituted phenylpyrazole with different electron-donating or electron-accepting groups. Photochem Photobiol Sci 20, 1487–1495 (2021). https://doi.org/10.1007/s43630-021-00125-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-021-00125-8