
Abstract
We report the structural properties, infrared (IR) and Raman spectra, dipole moment, polarisability, hardness and chemical potential of the trans and cis configurations of 4-hydroxyazobenzene calculated using the B3LYP functionals. All calculations were performed with the following basis sets: 6–31G, 6–31++G, 6–31G(d,p), 6–31++G(d,p), 6–31G(2d,2p), 6–31++G(2d,2p) and 6–311++G(2d,2p). We observed that 6–31++G(d,p) gives similar results to 6–311++G(2d,2p). Consequently, SVWN and PW91 methods were also used in association with 6–31++G(d,p) to test the influence of the different models of exchange and correlation functionals. A planar structure was obtained for all the optimised trans configuration structures. In both isomers, the presence of the hydroxyl group leads to an asymmetry in certain structural parameters. From these results, two IR or Raman active frequencies can be used to easily distinguish trans and cis configurations. The trans configuration was found to be more stable than the cis configuration by 67 ± 2 kJ mol−1 at 0 K. The difference of the dipole moment between trans and cis for 4-hydroxyazobenzene was found to be lower than for trans and cis azobenzene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Due to the difference in optical properties of the cis and trans isomer states, the industrial applications of azobenzene and derivatives could be considerable [1]. Hence, the number of theoretical studies has increased considerably during the last decade. Among the properties of interest, spectroscopic characterisation [2, 15] and cis-trans isomerisation mechanisms [16, 20] have been the most studied in the framework of density functional theory (DFT) using localised basis sets. The trans azobenzene (t-az) structure has been most studied from the experimental as well as the numerical point of view. The controversy about the planar structure of t-az seems resolved since numerical results [5, 13] were found to be in agreement with the most recent gas electrons diffraction (GED) experiment dating from 2001 [21]. However, the contradictory results obtained with and within the different methods serve to illustrate not only the difficulties of characterisation of this type of compound, but also the complementarities of numerical and experimental investigations.
Among the azobenzene derivatives 4-hydroxyazobenzene (OHaz), also known as phenlylazophenol, is of considerable industrial interest. Indeed, it can be used alone as a dye chromogen [22] or grafted by condensation to synthesise a photoresponsive polymer [23]. However, the number of experimental as well as numerical studies is limited. From the experimental point of view, only the trans configuration (t-OHaz) in its crystalline state has been characterised by X-ray diffraction [24] and had its vibrational spectra measured at different pH values [25]. The metastable nature of c-OHaz, which leads to difficult characterisation, explains why, to our knowledge, no data exist about this configuration. Theoretical studies have also only focused on t-OHaz. Indeed, Kurita et al. [27], using PW91/6–31+G**, studied the dimerisation of t-OHaz to explain its role in the photoisomerisation process. The absorption maximum of t-OHaz, among 43 azobenzene derivatives, was then evaluated by Chen et al. [3] using the ZINDO/S, CIS and TD methods using the B3LYP/cc-pVDZ optimised structure.
For photoresponsive applications, correct characterisation of both trans and cis configurations is required. This is the reason why, in this work, we used DFT in combination with different basis sets to characterise both t-OHaz and c-OHaz. The structural properties are compared to experimental results, where available, or to previous results obtained for azobenzene. The vibrational frequencies, IR and Raman intensities are discussed for the two configurations. Finally, electronic properties, including the energy, dipole moment, static polarisability, chemical potential and hardness, are presented. These results will be used to evaluate, among the tested basis sets, which is the most time-saving to describe the influence of the hydroxyl group on the azo bond features.
Methods
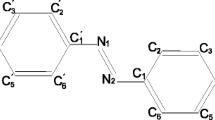
The atom numbering used in this study for 4-hydroxyazobenzene is presented in Fig. 1. Calculations were carried out using the GAUSSIAN98 [28] program with the DFT method. In the exchange correlation zoo, we choose the SVWN [29], PW91 [30, 32] and B3LYP [28, 33, 34] functionals. The energies of the structures were calculated with a SCF convergence on the density matrix of 10−8 a.u. All structures were optimised using the Berny algorithm [35] with the criteria for convergence being a maximum force less than 45×10−5 a.u and an rms force less than 3×10−4 a.u. Dunning’s correlation consistent basis set would have been the most suitable due to the fact that it is a correlation consistent basis set. However, the cc-pVTZ basis set generates 590 basis functions for 4-hydroxyazobenzene in comparison to 326 for 6–31++G(d,p) or 505 for 6–311++G(2d,2p). This increment of the basis functions means an increment of the time of calculation but also of the degrees of freedom during the self consistent field resolution, which can lead to numerical instabilities. To solve this, an improvement of numerical accuracy is sometimes required, consequently increasing the time of calculation and the memory space required. Fortunately, comparing the MP2/cc-pVTZ results with DFT results in the 6–31G*, Hättig et al. [5] observed that the differences for structural data are small compared with experimental uncertainties. Consequently, we tested the influence of diffusion and polarisation functions at the 6–31G level: 6–31G++, 6–31G(d,p), 6–31G(2d,2p), 6–31G++(d,p), 6–31G++(2d,2p) on the different properties in association with the B3LYP functional. Calculations at B3LYP/6–311G++(2d,2p) level were then performed and used to reference our results. For the two other functionals, the calculations were performed at the 6–31++G(d,p) level. Moreover, semiempirical methods with AM1 and PM3 models were also examined.

Trans and cis configuration of 4-hydroxyazobenzene.Black atoms Nitrogen, dark grey atoms carbon, white atoms hydrogen, light grey atoms oxygen. The axis reference is used to evaluate the polarisability tensor components
The IR absorption spectra were obtained with the intensities evaluated by the double harmonic approximation from dipole derivatives with respect to normal coordinates. Moreover, a finite field of 0.0019 a.u. was used to evaluate the Raman activities by differentiation of the polarisability with respect to the nth normal mode. The elements of the polarisability tensor are the second order response of the total energy with respect to a variation in the electric field. An analytic second derivative was obtained with the coupled-perturbed Kohn-Sham (CPKS) equations [36]. The mean value and the anisotropic polarisability were extracted from the polarisability tensor with the principal axis given in Fig. 1.
The chemical potential (μ) and absolute hardness (η) were evaluated by the finite-difference approximation from the ionization potential (I) and electron affinity (A) of the molecules. The last occupied Kohn-Sham orbital energy can be equated with the energy of the highest occupied molecular orbital (HOMO) associated to -I. Moreover, the first unoccupied Kohn-Sham orbital energy is often [37, 38] used as the value for the energy of the lowest unoccupied molecular orbital (LUMO) and so can be equated with -A even though it is a crude approximation since the DFT is a ground state theory.
Results and discussion
Structure
The characteristic geometric parameters of optimized t-OHaz and c-OHaz configurations obtained with different methods are given in Table 1. The X-ray measurements obtained for the crystalline structure of 4-hydroxyazobenzene [24] and the most recent GED and X-ray data concerning azobenzene [21, 39, 40] have also been included. Semiempirical methods have been extensively used to study azobenzene derivatives [41, 44] in the last decade. The semiempirical data obtained for t-OHaz are in good agreement with the experimental and DFT results, while the discrepancies are important in the case of c-OHaz. Our results are similar to those presented by Kurita et al. [12] for t-OHaz. However, for c-OHaz, the discrepancies with DFT results are larger than those observed for c-az, mainly for bending and dihedral angles. Consequently, the results seem to confirm that a semiempirical optimised geometry has to be studied carefully, especially for the cis configuration. Concerning the basis set dependence of the structural parameters, we noted that, as expected, the inclusion of diffuse and polarisation functions leads to a large change in the optimised structures. Addition of diffuse functions slightly increases both bond lengths and angles whereas polarisation functions drastically decrease these values.
Indeed, for both trans and cis configurations, we noted that 6–31G++ yields a N1–N2 bond length 1.4% shorter than 6–31++G(d,p). However, the most important effect concerns the C2–N1–N2–C3 and N2–N1–C2–C1 angles of the cis configuration, with a relative difference of 8% between the two methods. From 6–31++G(d,p) to 6–311++G(2d,2p) the numerical uncertainty is in general around 0.2 pm for the bond lengths and less than 1° for the angle. Consequently, from 6–31++G(d,p) experimental and numerical uncertainty is similar [11]. Thermal effects are one of the main causes for the discrepancy between numerical and experimental results. Indeed, DFT calculations are performed at 0 K whereas experimental measurements are performed at higher temperatures. This effect is important since, by X-ray measurement [39], N = N bond lengths were measured at 1.259 Å and 1.249 Å at 82 K and 296 K, respectively. The same effect was observed for the C–N = N angle, with values of 114.1° and 113.5° for 82 K and 296 K, respectively.
The calculated bond distances, as well as bond angles, using local (SVWN) and nonlocal (PW91) functionals show some differences from the results obtained with a hybrid functional (B3LYP). In general, the local functionals tend to favour the area of high electron density, resulting in short bond distances, whereas non-local functionals tend to overcorrect this effect. This effect is noticeable on both the t-OHaz and c-OHaz optimised geometries since, in general, the N=N distance is shorter and longer compared to B3LYP/6–31G++(d,p), respectively, for SVWN/6–31G++(d,p) and PW91/6–31G++(d,p). Only N=N of t-OHaz obtained for B3LYP/6–31G++(d,p) and SVWN/6–31G++(d,p) are of the same order, otherwise the results deviate by 2 pm. Concerning the angles, the choice of the functional does not drastically affect the bond angles, whereas the deviations are larger for the dihedral angles. Indeed both SVWN and PW91 overestimate the C2–N1–N2–C3 dihedral angle, whereas N1–N2–C3–C4 and N2–N1–C2–C1 are underestimated in comparison with B3LYP results. These conclusions about the functionals are in agreement with previous results of Biswas et al. [2] on azobenzene.
Compared with previous results obtained for azobenzene, we noted that the presence of the hydroxyl group induces an asymmetry of the two configurations. Indeed, the N1–C2 bond length is slightly higher than the N2–C3 bond lengths. This asymmetry was observed by Kurita and al. [14] using PW91/6–31+G(d), but only on the trans configuration and it was absent from X-ray measurements [24]. The asymmetries were also observed in the N–N–C–C torsion angles in the cis configuration, since the N1–N2–C3–C4 torsion angle was systematically lower than that of N2–N1–C2–C1. These asymmetries are independent of the method of calculation and can be attributed to a physical effect due to the presence of the activating hydroxyl group.
It is also worth noting that, compared to previous numerical results obtained for t-az, we obtained a planar structure for all the optimised trans structures. Consequently, these results illustrate the fact that, in addition to model dependent parameters such as method, functionals or basis set, the numerical parameters such as convergence criteria or numerical integrations precision are also crucial for the accuracy of the calculated properties.
Frequency
The Raman and infrared (IR) spectra of trans and cis 4-hydroxyazobenzene obtained with 6–311++G(2d,2p) are presented in Figs. 2 and 3. The harmonic frequencies, without scaling factor, of selected modes will be compared to available experimental data obtained for t-OHaz [25]. Moreover, the vibrational properties of azobenzene were numerically [2, 8, 9, 12] extensively studied and compared with experimental data [45–47]. In general, from 6–31G(d,p) and beyond, the wavenumbers are known with an error not exceeding 15 cm−1 for 67 modes out of 69. However, the choice of the basis set affects the activity of the modes. Moreover, compared to the B3LYP/6–31G++(d,p) method, the calculated modes at PW91/6–31G++(d,p) and SVWN/6–31G++(d,p) levels are overestimated. The IR activities evaluated by the three functionals are qualitatively comparable, but this is not the case for the Raman activities.

6–311++G(2d,2p) Raman (a) and infrared (IR) (b) spectra of trans 4-hydroxyazobenzene. The calculated harmonic frequencies without scaling factor are represented with a Gaussian bandwith of 10 cm−1

6–311++G(2d,2p) Raman (a) and IR (b) spectra of cis 4-hydroxyazobenzene. The calculated harmonic frequencies without scaling factor are represented with a Gaussian bandwith of 10 cm−1
The inclusion of diffuse functions particularly affects the modes evaluated, independent of the configuration, at 351 cm−1 and 1,282 cm−1 with 6–311++G(2d,2p). The reduction of the basis set at 6–31G(2d,2p) caused a blue shift of 20 and 27 cm−1, respectively, for these two modes. The same results were obtained for 6–31G(d,p). The mode at 351 cm−1 is assigned to the OH out-of-plane bending, and the mode at 1,282 cm−1 to CO stretching and CC and CH in-plane bending. These modes are among the most IR active and are clearly visible in the spectra in Figs. 2b and 3b. This is also the case for the modes at 1,644 cm−1 (CC stretching and CH in-plane bending), and the mode at 1,196 cm−1 (OH in-plane bending). All these modes are independent of the configuration. The N = N stretching modes of trans azobenzene derivatives were in general measured between 1,380 cm−1 and 1,460 cm−1, and precisely at 1,442 cm−1 for t-OHaz. From the present 6–311++G(2d,2p) results, N = N stretching is implied in five modes calculated at 1,467 cm−1, 1,483 cm−1, 1,506 cm−1, 1,526 cm−1 and 1,544 cm−1. The values of the modes are relatively basis-set-independent, with a general shift of ±10 cm−1 as a function of the modes studied and the basis set used. However, this is not the case with activity, since the mode at 1,467 ± 10 cm−1 or at 1,541 ± 10 cm−1 is very sensitive to the choice of the basis set and of the functionals. This can explain why for azobenzene the value of N = N stretching mode was obtained over the range from 1,420 cm−1 to 1,556 cm−1 by different authors [2, 8, 9, 12]. From the B3LYP/6–311+G(2d,2p) calculation, the N = N stretching mode with the highest activity is evaluated at 1,591 cm−1 for c-OHaz. This mode corresponds to the mode evaluated at 1,544 cm−1 for t-OHaz. Consequently, a blue shift of 46 cm−1 appears between t-OHaz and c-OHaz. This shift ranges from 38 to 57 cm−1 as a function of the basis set from the 6–31G(d,p) model, whereas it was evaluated at 64 and 73 cm−1, respectively, for 6–31++G and 6–31G. For azobenzene, the experimental value is 68 cm−1 and was numerically evaluated at 50 cm−1 using MP2/cc-pVTZ [11], 43 cm−1 using B3LYP/6–31+G(d,p) [8] and 92 cm−1 using BP86/6–31G* [2] and BP86/TZVP [11]. Consequently, our results are similar to previous results obtained for azobenzene with the same functional indicating that the hydroxyl group does not affect this mode. Concerning the effect of the functional, the blue shift is reproduced by the three functionals, but with PW91/6–31G++(d,p) it is only of 15 cm−1 compare to 38 cm−1 and 44 cm−1, respectively, obtained for B3LYP/6–31G++(d,p) and SVWN/6–31G++(d,p).
The C–N symmetrical stretch mode was experimentally assigned to a strong band at 1,147 cm−1. From B3LYP/6–311++G(d,p) results, this mode is assigned at 1,156 cm−1 and 1,140 cm−1, respectively, for t-OHaz and c-OHaz. This shift of 16 cm−1 is consistent with that of 20 cm−1 calculated by Biswas et al. [2] for azobenzene. At the B3LYP/6–31++G(d,p) and PW91/6–31++G(d,p) level, this shift is 14 cm−1, but at the SVWN/6–31++G(d,p) level we note an inversion, with the modes assigned at 1,114 cm−1 and 1,128 cm−1, respectively, for t-OHaz and c-OHaz. Moreover, the Raman activity of this mode predicted by the SVWN/6–31++G(d,p) method is abnormally weak compared to the other methods. The phenyl CH bending modes measured at 1,187 cm−1 and 1,318 cm−1, and the CC stretching modes measured at 1,467 cm−1 and 1,602 cm−1 are also characteristic of the experimental Raman spectrum. Three out of four of these modes are clearly visible as single peaks in Fig. 2a at 1,208 cm1, 1,339 cm−1 and 1,644 cm−1 whereas the last one is calculated at 1,282 cm−1 with a very low activity. The SVWN/6–31++G(d,p) method overestimates these modes this is particularly true for the second mode evaluated at 1,424 cm−1. On the contrary, these modes are well reproduced by the PW91/6–31+G(d,p) method, with the values calculated as 1,184 cm−1, 1,269 cm−1 1,369 cm−1 and 1,611 cm−1.
Consequently, compared to measured characteristic bands of azobenzene, the precision of the results are approximately the same using 6–31++G or 6–311++G(2d,2p). From the first to the last tested model, a blue shift of 30 cm−1 can be observed for certain modes. The relative intensities of these shifted modes being extremely low, the spectrums are not much affected. However, the improvement in Raman relative intensities induced by polarisation functions allows the visualisation of bands characteristic of 4-hydroxyazobenzene for trans configuration. Concerning the choice of functionals, the IR spectra obtained for the three functionals are qualitatively comparable both for the cis and the trans configuration. However, the difference of the N = N stretching mode between t-OHaz and c-OHaz calculated at PW91/6–31+G(d,p) seems underestimated.
Electronic properties
The total energy, zero point energy and entropy of both trans and cis configurations are given in Table 2. As expected, t-OHaz is always determined as the most stable configuration. From 6–31G(d,p) and beyond, t-OHaz is more stable than c-OHaz by 67 ± 2 kJ mol−1, whereas the relative stability is more important for poorer basis sets. The values obtained for SVWN/6–31G++(d,p) and PW91/6–31G++(d,p) models are lower with 61 kJ mol−1 and 63 kJ mol−1, respectively. For azobenzene, the theoretical values are in the range of 43–66 kJ mol−1, whereas both combustion [48] and photocalorimetry [49] experiments measured a value of 50.16 kJ mol−1. Thus, we can assume that polarisation functions correct the overestimation of the relative stability. Contrary to the entropy, the relative difference of the zero point energy is not basis-set-dependent. The shift of the frequencies induced by the inclusion of the polarisation functions affects mainly the low frequencies modes of the t-OHaz. Consequently, the entropy of t-OHaz evaluated by 6–31G is 8 J/mol K higher than with 6–311++G(d,p). For c-OHaz, the low frequency modes are less affected and the difference in entropy evaluated from the two extreme basis sets is only 4 J/mol K. Consequently, the entropy evaluated by 6–31G and 6–31++G calculations on c-OHaz is about 3 J/mol K greater than t-OHaz, whereas from 6–31G(d,p) and beyond, the entropy of t-OHaz is higher than for c-OHaz.
The dipole moment (D), mean(<α>) and anisotropic([α]a) polarisability, HOMO, LUMO, chemical potential (μ) and hardness (η) are of considerable interest for the design of light-driven molecular switches [50]. The values evaluated with different basis sets are given in Table 3. As expected, the molecular dipole moment is basis set- and functionals-dependent. In general, the value of the dipole moment decreases with the size of the basis set, whereas the values evaluated with the PW91/6–31G++(d,p) and SVWN/6–31G++(d,p) methods are slightly higher than those obtained with the B3LYP/6–31G++(d,p) method. However, these shifts are similar in c-OHaz and t-OHaz. Consequently, the difference in dipole moment between the two configurations is around 2.5 ± 0.2 D except for the B3LYP/6–31G and B3LYP/6–31G++ methods, where the values are around 3 D. For azobenzene, this difference was measured at 3 D [51] and calculated at 3.2 D and 3.3 D with the RPBE/DNP [52] and B3LYP/6–31+G(d,p) [8] methods, respectively. With the B3LYP/6–31G++(d,p) method, the value found was 3.36 D. In comparison with the results obtained for B3LYP/6–31++G(d,p) we can see that the hydroxyl group increases the dipole moment of t-OHaz by 1.34 D and by only 0.83 D for c-OHaz. Consequently, the difference in dipole moment between the trans and cis configurations of 4-hydroxyazobenzene is far lower than in the case of azobenzene. We noted that AM1 results are in correct agreement with those calculated with DFT despite discrepancies concerning the structural parameters of the cis configuration. The same result is obtained for static polarisability. The difference of mean polarisability calculated with the B3LYP method between c-OHaz and t-OHaz is converged around 26 ± 2 a.u.,whereas the net values are basis-set-dependent. This value is around 5 a.u lower than those evaluated with the SVWN/6–31G++(d,p) and PW91/6–31G++(d,p) methods. For t-az, the value has been evaluated at 183 a.u by the B3LYP/6–31G++(d,p) method in agreement with the value obtained by Hinchliffe et al. [54] and at 160 a.u. for c-az. It is commonly stated that, at equilibrium geometry, a molecule possesses a maximum hardness and a minimum polarisability in comparison with other configurations. These principles are known as the maximum hardness principle (MHP) [54] and minimum polarisability principle (MPP) [54]. Although extensively used, these principles are still debated [37]. From our results, t-OHaz is the stable structure for all the basis sets, and its hardness is always higher than that of c-OHaz, hence the MHP is verified. However, higher polarisability is obtained for t-OHaz independently of the basis set and of the functionals used, hence MPP is not valid. Several factors can lead to this result. The first could be due to the fact that different isomers exist as a function of hydroxyl group configuration, and that the configuration used in this work was not the most stable. Secondly, the reason could be the numerical parameters used in the calculation of the polarisability tensor, including the functional, the intensity of the electric field and the others parameters used in the determination of the derivative of the density matrix with respect to the electric field. Another reason could be the inability of the DFT method to well predict the polarisability of the system. Finally, a breakdown of the MPP principle in π-conjugated molecules could be involved, as already observed by Torrent-Sucarrat [38, 56]. Consequently, a more complete study would be necessary before a global conclusion can be made about the application of MPP and MHP to 4-hydroxyazobenzene.
Conclusions
From these results it appears that the optimised structural parameters predicted by semiempirical methods are in correct agreement with DFT results concerning the trans configuration, whereas the optimised geometry of the cis configuration must be analysed carefully. Concerning the DFT results, the structural parameters are determined within acceptable precision for the 6–31G(d,p) basis set and beyond. Looking at the frequencies of the characteristic modes of the azobenzene group, the precision of the results are approximately the same using the 6–31++G or 6–31++G(2d,2p) sets compared to experimental results. Compared to Raman experimental data, the main characteristic modes of the trans configuration were well described by the B3LYP/6–311++G(2d,2p) method. However, the Raman activities of these modes are very basis set- and functional-sensitive. The PW91 functional underestimates the shift of the N = N stretching mode between the trans and cis configuration. Concerning electronic properties, the accuracy obtained with the 6–31++G(d,p) basis set is correct compared to 6–311++G(2d,2p) for polarisation, HOMO and LUMO energies. Nevertheless, this is not the case for the dipole moment, with a discrepancy of 3% between the two methods.
Concerning the properties of 4-hydroxyazobenzene, planar trans optimised structures were obtained with all the basis sets and functionals. An asymmetry in the structural parameters was observed on both structures. The presence of the hydroxyl group does not shift the characteristic modes of the azo group. The trans configuration was found to be more stable than the cis configuration by 67 ± 2 kJ mol−1. The relative difference of the dipole moment between the trans and cis configurations was found to be lower for 4-hydroxyazobenzene than for azobenzene. However, a more complete study is necessary to obtain the statistical properties of 4-hydroxyazobenzene since the presence of the hydroxyl group induces more degrees of freedom than in azobenzene.
References
Halabieh RHE, Mermu O, Barrett C (2004) J Pure Appl Chem 76:1445–1465
Biswas N, Umapathy S (1997) J Phys Chem A 101:5555–5566
Chen PC, Chieh, YC, Wu JC (2005) J Mol Struct 715:183–189
Chen PC, Chieh YC (2003) J Mol Struct 624:191–200
Hättig, Hald K (2002) Phys Chem Chem Phys 4:2111–2118
Lednev IK Ye TQ, Matousek P, Towrie M, Foggi P, Neuwahl FVR, Umapathy S, Hester RE, Moore JN (1998) Chem Phys Lett 290:68–74
Liu J, Chen Z, Yuan S (2005) Zhejiang Univ SCI 6B:584–589
Stepanic V, Baranovic G, Smrecki V (2001) J Mol Struct 569:89–109
Armstrong DR, Clarkson J, Smith WE (1995) J Phys Chem 99:17825–17831
Cattaneo P, Persico M (1997) Phys Chem Chem Phys 1:4739–4743
Fliegl H, Köhn A, Hättig C, Ahlrichs R (2003) J Am Chem Soc 125:9821–9827
Kurita N, Tanaka S, Itoh S (2000) J Phys Chem A 104:8114–8120
Briquet L, Vercauteren DP, Perpete EA, Jacquemin D (2006) Chem Phys Lett 417:190–195
Kurita N, Ikegami T, Ishikawa Y (2002) Chem Phys Lett 360:349–354
Jha PC, Pati YA, Ramasesha S (2005) Mol Phys 103:1859–1873
Tiago ML, Ismail-Beigi S, Louie SG (2005) J Chem Phys 122:094311–0943171
Kucharski S, Janik R, Motschmann H, Radüge C (1999) New J Chem 23:765–771
Zheng X, Sohlberg K (2004) J Comp Theor Nano 1:187–192
Cembran A, Bernardi F, Garavelli M, Gagliardi L, Orlando G (2004) J Am Chem Soc 126:3234–3243
Kikuchi O, Azuki M, Inadomi Y, Morihashi K (1999) J Mol Struct 468:95–104
Tsuji T, Takashima H, Takeuchi H, Egawa T, Konaka S (2001) J Phys Chem A 105:9347–9353
Mishima K, Matsuyama K, Ishikawa H, Hayashi K, Maeda S (2002) Fluid Phase Equilib 194–197:895–904
Gan LH, Xia X, Chan CY, Hu X, Zhao X (2003) Polymer Adv Tech 14:260–265
Shamuratov EB, Batsanov AS, Struchkov YT, Shukurov A, Makhsumov AG, Sabirov VK (1991) J Struct Chem 32:447–449
Uno T, Lee H, Saito Y, Machida K (1976) Spectrochim Acta A 32:1319–1322
Tecklenburg MJ, Kosnak DJ, Bhatnagar A, Mohanty DK (1997) J Raman Spectrosc 28:755–763
Kurita N, Nebashi S, Kojima M (2005) Chem Phys Lett 408:197–204
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA (1998) Gaussian 98, Revision A7. Gaussian, Pittsburgh, PA
Vosko SH, Wilk L, Nusair M (1980) Can J Phys 58:1200–1211
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Phys Rev B 46:6671–6687
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1993) Phys Rev B 48:4978–4978
Adamo C, Barone V (1997) Chem Phys Lett 274:242–250
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Schlegel HB (1982) J Comp Chem 3:214–218
Fournier R, Andzelm J, Salahub DR (1989) J Chem Phys 90:6371–6377
Ghanty TK, Ghosh SK (1996) J Phys Chem 100:12295–12298
Torrent-Sucarrat M, Luis JM, Duran M, Sola M (2001) J Am Chem Soc 123:7951–7952
Harada J, Ogawa K, Tomoda S (1997) Acta Crystallogr B 53:662–672
Mostad A, Romming C (1971) Acta Chem Scand 25:561–564
Shaabani A, Zahedi M (2000) J Mol Struct (Theochem) 506:257–261
Muzikante I, Gerca L, Fonavs E, Rutkis M, Gustina D, Markava E, Stiller B, Brehmer L, Knochenhauer G (2002) Mater Sci Eng C 22:339–343
Weber R, Winter B, Hertel IV, Stiller B, Schrarder S, Brehmer L, Koch N (2003) J Phys Chem B 107:7768–7775
Manaka T, Shimura D, Iwamoto M (2002) Chem Phys Lett 355:164–168
Kellerer B, Hacker HH, Brandmuller J (1971) Ind J Pure Appl Phys 9:903–907
Kubler VR, Luttke W, Weckherlin SZ (1960) Elektrochem 64:650–654
Smercki V, Baranovic G, Keresztury G, Meic Z (1997) J Mol Struct 408/409:405–408
Corrucini RJ, Gilbert EC (1939) J Am Chem Soc 61:2925–2927
Adamson AW, Vogler A, Kunkely H, Wachter R (1978) J Am Chem Soc 100:1298–1300
Zhang C, Du MH, Cheng HP, Zhang XG, Roitberg AE, Krause JL (2004) Phys Rev Lett 92:158301–1–158301–4
Kumar GS, Neckers DC (1989) Chem Rev 89:1915–1925
Tong X, Wang, G, Soldera A, Zhao Y (2005) J Phys Chem B 109:20281–20287
Hinchliffe A, Nikolaidi B, Machado HJS (2004) Int J Mol Sci 5:224–238
Pearson RG (1987) J Chem Educ 64:561–567
Chattaraj PK, Sengupta S (1996) J Phys Chem 100:16126–16130
Torrent-Sucarrat M, Luis JM, Solà M (2005) Chem Eur J 11:6024–6031
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Minisini, B., Fayet, G., Tsobnang, F. et al. Density functional theory characterisation of 4-hydroxyazobenzene. J Mol Model 13, 1227–1235 (2007). https://doi.org/10.1007/s00894-007-0244-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-007-0244-1