
Abstract
Monte Carlo simulations of the single- and double-walled carbon nanotubes (CNT) intercalated with different metals have been carried out. The interrelation between the length of a CNT, the number and type of metal atoms has also been established. This research is aimed at studying intercalated systems based on CNTs and d-metals such as Fe and Co. Factors influencing the stability of these composites have been determined theoretically by the Monte Carlo method with the Tersoff potential. The modeling of CNTs intercalated with metals by the Monte Carlo method has proved that there is a correlation between the length of a CNT and the number of endo-atoms of specific type. Thus, in the case of a metallic CNT (9,0) with length 17 bands (3.60 nm), in contrast to Co atoms, Fe atoms are extruded out of the CNT if the number of atoms in the CNT is not less than eight. Thus, this paper shows that a CNT of a certain size can be intercalated with no more than eight Fe atoms. The systems investigated are stabilized by coordination of 3d-atoms close to the CNT wall with a radius-vector of (0.18–0.20) nm. Another characteristic feature is that, within the temperature range of (400–700) K, small systems exhibit ground-state stabilization which is not characteristic of the higher ones. The behavior of Fe and Co endo-atoms between the walls of a double-walled carbon nanotube (DW CNT) is explained by a dominating van der Waals interaction between the Co atoms themselves, which is not true for the Fe atoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Investigations of nanosized carbon clusters are of great interest because of their relevance to the creation of new materials in electronics [1], biology, chemistry, etc. The last decade saw the modeling of constituent elements and processes related to carbon cluster/intercalate systems: interaction of carbon fullerenes and nanotubes with hydrocarbons and their derivatives [2] and the adsorption by fullerenes of different aromatic hydrocarbons, alcohols, etc. from gas and liquid phases [3]. We have also reported calculations by the Monte-Carlo method and the methods of molecular dynamics of lithium and hydrogen adsorption [4, 5]. Paper [6] describes magnetic properties of the Fe1−xNix mixture added to carbon nanotubes.
The research reported here deals with intercalation of single- and double-walled carbon nanotubes by such metals as Fe or Co. A comparison of metal/metal interaction and metal/carbon (nanotube wall) interactions allows us to determine the predominant value of carbon/metal interactions which is indirectly confirmed by other work of the authors [7].
Materials and methods
All calculations were carried out using the computer Monte-Carlo method. Models of single-walled “zigzag” nanotubes, each consisting of 324 carbon atoms and having 0.68 nm diameter and 3.64 nm length were assembled. Inside were placed 8 metal atoms at a distance of the effective interaction radius, which did not exceed 0.14 nm for Fe and 0.135 nm for Co.
In the model considered, the potential given in Reference [8] was combined with the Born–Mayer repulsive pair potential that describes high-energy atoms collisions [9] within the range 0–0.14 nm. Interaction of carbon atoms is described by the Tersoff–Brenner multi-particle potential with a radius of 0.21 nm [10] combined with the Zigler–Birzak–Litmark repulsive pair potential [9]. The distance between the carbon atoms was 0.139 nm, and between the walls (in double-walled nanotubes) — 0.335 nm. The Fe–C and Co–C interactions within the range of 0.195–0.375 nm were described by a Lennard–Jones pair potential [11] with minimum Carbon–metal interaction energy of−0.11 eV at an interatomic distance of 0.235 nm. The modeled period of development of one cascade of collisions was 2 ps, and the energy conservation law in every model cascade was observed to within 0.1%. Initial coordinates of metal atoms inside a single-walled nanotube and within the interwall distance in a double-walled nanotube were selected according to the law of random numbers.
Results and discussions
Single-walled CNT
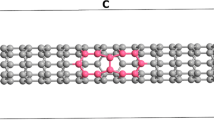
To define general and constant calculation conditions, a metallic single-walled carbon nanotube (SW CNT) (9,0) with the side surface length equal to 17 bands was selected. This structure consists of benzene rings (Fig. 1), since SW CNT length reduction gives the unfavorable effect of extruding endo-atoms from the cylinder, even at low temperatures.

Metallic single-walled carbon nanotube structure consisting of benzene rings
The temperature range varied from 0 to 900 K, to exclude the loss of data relative to the stability of the intercalated system, which was considered in a vacuum. However, of special interest will be the calculation results in the range of (300–800) K, because no such peculiarities have been revealed at lower temperatures. Temperatures over 800 K may result in destruction and/or sublimation of the organic carbon phase. Moreover, a cell was selected that allows for endo-atom oscillation of about 1 nm, which, on the one hand, allows for “free” atomic motion along all the spatial directions, and, on the other hand, does not let the SW CNT extrude the atom easily out of the system. Energy parameters were defined within 0.001 J mol−1 with the heating step of 1 K.
Our research has proved the systems behave similarly for the cases of one, two and three Fe endo-atoms (see Fig. 2).

One, two and three Fe endo-atoms
We can see that systems 2 and 3 are more stable than systems 1 and 4. Besides, each system has its own stabilization conditions: 1 — (450–600) K; 2 — (380–550) K; 3 — (430–550) K and 4 — (400–800) K. This means that systems 2 and 3 are in the course of stabilization related to the formation of a coordination interaction between the SW CNT π-shell and free Fe d-orbitals, which is not found for system 4, in which the interaction of Fe atoms dominates. Of special interest is the case with system 4, which is to some extent “saturated” (temperature range is (600–750) K). Within such a range, this system turns into a system consisting of a free SW CNT only, with four Fe atoms almost strictly parallel to the main axis. The total energy of such an intercalated system becomes slightly lower, mostly due to the coordination interaction. An example of such a structure is given in Fig. 1.
Systems containing five (structure 5), six (structure 6), seven (structure 7) and eight (structure 8) Fe endo-atoms are unique: structures 5, 6 and 7 have similar energy parameters. Besides, their stabilities are also similar, they do not depend on the number of endo-atoms. Structure 8 has a lower energy than the three previous ones, but at about 800 K it behaves similarly, extruding one Fe atom in two opposite directions — to the open ends of SW CNT. The extrusion result is shown in Fig. 3. The energy of system 8 does not decrease.

Extrusion result of one Fe atom in two opposite directions — to the open ends of SW CNT
Thus, a coordination interaction of the endo-atom with the π-shell of the SW CNT takes place in all the cases investigated. The interaction distances are (0.20–0.25±1) nm. Moreover, if the number of endo-atoms is not less than eight, the extrusion process is regular if the SW CNT length does not exceed 17 bands.
For comparison, SW CNTs intercalated with Co atoms were also investigated. An interesting result is observed with systems 1 and 2 (see Fig. 4). Their stabilities are close to those of systems 4, 6 and 7 with Fe. However, in the range of (440–455) K (peak on curve 1, Fig. 4) we have partial stabilization of system 1 due to a coordination interaction of all Co endo-atoms with the side surface of SW CNT, with a radius-vector of (0.18–0.20) nm.

SW CNTs intercalated with Co atoms in systems 1 and 2
With increased intercalate concentration inside the SW CNT, nothing special occurs that might influence the system’s geometric parameters. There is no extrusion of Co endo-atoms out of the SW CNT, even in the case with eight atoms.
Double-walled CNT
Quite another picture is observed with a double-walled carbon nanotube (DW CNT) intercalated by Fe and Co atoms (Fig. 5).

A double-walled carbon nanotube (DW CNT) intercalated by Fe and Co atoms
Calculation of such a system required an approximation of singling out a small nanotube fragment from the three layers while maintaining the intercalate concentration proportions with respect to a fixed nanotube length. Thus, a system of a coaxial metallic DW CNT (9,0)/(19,0) with an inter-wall distance of 0.38 nm was investigated. Similarly to the previous discussions concerning the model design, Fe and Co atoms were placed in the internal cylinder SW CNT (9,0) of a DW CNT along its rotation axis. However, the intertubular space was filled to take into account that the arrangement of metal atoms in the plane perpendicular to the DW CNT axis is less stable (Fig. 5).
Thus, the balance conditions between the necessary spontaneous smoothing of the system being heated and the dispersion interaction forces between the walls of a DW CNT and its intercalate were created artificially.
As mentioned above, double systems are characterized by a radically different tendency towards stabilization with respect to SW CNTs. This is manifested in more complicated endo-atom extrusion. According to Fig. 6, Fe atoms of SW CNT (9,0) show stronger dispersion interactions compared to a similar SW CNT (9,0), which results in their aggregation inside the cylinder or in weak extrusion. Accordingly, Fe atoms are coordinated near the open CNT end, their concentration being increased (Fig. 6).

Fe atoms of SW CNT (9,0) showing stronger dispersion interactions compared to a similar SW CNT (9,0)
The tendencies observed with SW CNTs are also characteristic, though to a lesser extent, of the system of coaxial DW CNTs with Co endo-atoms. However, an essential distinction in the behavior of SW and DW CNTs is the pole difference in the extrusion of Fe and Co atoms from the SW CNT (9,0) of the DW CNT (9,0)/(19,0) systems. This means that if SW CNTs show stronger interactions between Co atoms, and Fe atoms can easily be moved out of the CNT at high temperature, then the DW CNTs (9,0)/(19,0) display quite different behavior: full extrusion of Co endo-atoms from the SW CNT (9,0) (Fig. 7).

DW CNTs (9,0)/(19,0) displaying full extrusion of Ñî endo-atoms from the SW CNT (9,0)
An interesting specific interaction is observed with the atoms placed between the walls of two coaxial CNTs (9,0)/(19,0). The calculation suggests that it is better to form dispersion distances between the metal atoms themselves than between a metal and a CNT wall. This is true for both DW CNT systems — those with Co and Fe. However, there are some distinctions. When heated, a CNT system with eight Co atoms placed between the walls is quickly stabilized by pushing out a large number of these atoms (Fig. 8, Structure 1). Quite different behavior is observed with Fe (Fig. 8, Structure 2).

Stabilization of CNT system with eight Co atoms placed between the walls
Thus, the modeling of CNTs intercalated with metals by the Monte Carlo method suggests that there is a correlation between the length of a CNT and a number of endo-atoms of a specific type. Thus, in the case of the CNT (9,0) of length 17 bands, unlike Co atoms, the extrusion of the Fe atom out of the CNT is observed if the number of atoms in the CNT is not less than eight. Thus, with some degree of certainty we can state that this CNT can be intercalated with no more than eight Fe atoms by ionic impact or chemical modification. The systems investigated are stabilized by coordination of d-atoms close to the CNT wall, with a radius-vector of 0.18–0.20 nm. Another characteristic feature is that, within the temperature range of (400–700) K, small systems exhibit ground-state stabilization, which is not observed for the higher ones.
Thus, the calculations revealed a principal difference in intercalation of SW and DW CNTs which lies in diametrically opposite behavior of Fe and Co endo-atoms in an SW CNT.
The behavior of Fe and Co endo-atoms between the DW CNT walls is explained by a dominating van der Waals interaction between the Fe atoms themselves, which is not true for the Co atoms.
References
Axenov AI, Bobrinetskii II, Bulatov AN, Nevolin VK (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 255
Agafonov SS, Glazkov VP, Somenkov VA, Fillipov AA (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 254
Berezkin VI, Samonin VV, Nikonova VY (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 257
Fedorov AS, Sorokin PV (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 260
Fedorov AS, Sorokin PV (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 261
Wei X-W, Wu H-Q, Cao Y-J, Yuan P-S, Xu H-Y (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 270
Belova E, Chernozatonskii L (2005) International conferences “Fullerenes and Atomic Clusters”, St. Petersburg, Russia, p 256
Gades H, Urbassek HM (1992) Nukl Instr And Meth B 69:232–234
Rapaport DC (1995) The art of molecular dynamics simulation. Cambridge University Press, Cambridge, UK
Tersoff J (1989) Phys Rev 39:5566–5568
Dorfman S, Mundim KC, Fuks D, Berner A, Ellis DE, van Humbeeck J (2001) Mat Sci and Eng C 15:191
Acknowledgements
This work was supported by the CRDF grant (UKP1-2616-KV-04) and partly by the “Dnipro” Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mykhailenko, O., Matsui, D., Prylutskyy, Y. et al. Monte Carlo simulation of intercalated carbon nanotubes. J Mol Model 13, 283–287 (2007). https://doi.org/10.1007/s00894-006-0129-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-006-0129-8