
Abstract
The kinetics of the hydrogen abstraction reactions of hydrogen atom with n-propyl radical and isopropyl radical were studied using the direct ab initio dynamics approach. BHandHLYP/cc-pVDZ method was employed to optimize the geometries of stationary points as well as the points on the minimum energy path (MEP). The energies of all the points for the two reactions were further refined at the QCISD(T)/cc-pVTZ level of theory. No barrier was found at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory for both reactions. The forward and reverse rate constants were evaluated with both canonical variational transition state theory (CVT) and microcanonical variational transition state theory (μ VT) in the temperature range of 300–2,500 K. The fitted three-parameter Arrhenius expression of the calculated CVT rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory are kCVT (n – C3H7)=1.68×10−14 T0.84 e(319.5/T) cm3 molecule−1 s−1 and kCVT (iso-C3H7)=4.99×10−14 T0.90 e(159.5/T) cm3 molecule−1 s−1 for reactions of n-C3H7 + H and iso-C3H7 + H, respectively, which are in good agreement with available literature data. The variational effects were analysed.
Figure Comparison of the calculated forward rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory and the available experimental and theoretical data of the reaction vs 1,000/T for the two reactions.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The elementary reactions of alkyl radicals with hydrogen atom in the gas phase are of considerable importance in the course of combustion of hydrocarbons. Particularly, under pyrolytic conditions (in the absence of oxidants), hydrogen atoms can be the primary chain carriers in thermal decomposition of hydrocarbons. Due to the difficulties in experimental techniques, there are few of such elementary reactions where the experimental kinetic data are available. The major difficulty in experimental study is that the reactions between radicals occur much faster than those between neutral molecules. In such a case, theory can play an important role in providing necessary kinetics information.
The n-propyl radical and isopropyl radical are two important intermediate species in the atmosphere and in the course of combustion of hydrocarbons. The kinetics data of these reactions are essential for modeling the chemical process involved. The recently reported reaction class approach showed promising feature for predicting rate constants for bulk of reactions with the same reaction moiety. However, the applicability of the methods still needs validation to help further promote them. The reactions of propyl radical with hydrogen atom consist of two types of reactions: one is the combination reaction of propyl radical with hydrogen atom yielding propane; another is the hydrogen abstraction reaction of propyl radical with hydrogen atom yielding propylene and hydrogen molecule. The latter is the objective of this present study, which is of interest to us. The reactions of the present study are good prototype for the hydrogen abstraction reaction class of hydrogen atom with alkyl radicals, although the rate constant of the latter is slightly slower than that of the former. In spite of the importance in combustion and atmospheric chemistry, no direct measurement of the rate constants of the direct hydrogen abstraction reactions of hydrogen atom with n-propyl radical and isopropyl radical have been reported to the best of our present knowledge. In an extensive literature review, Tsang reported the rate constants of k=3×10−12 cm3 molecule−1 s−1 and k=6×10−12 cm3 molecule−1 s−1 using the bond energy and bond order (BEBO) method with an uncertainty factor of 2 over the temperature range of 300–2,500 K for n-C3H7 + H → C3H6 + H2 and iso-C3H7 + H → C3H6 + H2, respectively. However, these results were obtained based upon the relative rate constants to other reactions within a limited temperature range. Thus, high-level ab initio calculations and rate constant calculations are still required for accurate rate constants over a wide temperature range.
In the present study, our primary objective is to calculate kinetics data of the reactions that yield stable propylene, which in turn will be employed to predict rate constants for similar reactions from the reaction class transition state theory (RC-TST) proposed by T.N. Truong. In view of the above case, we first constructed the minimum energy paths (MEPs) for the direct hydrogen abstraction reactions of n-C3H7 + H → C3H6 + H2 and iso-C3H7 + H → C3H6 + H2. Then the direct ab initio dynamics method was adopted to investigate the dynamic properties of the two direct hydrogen abstraction reactions based on MEP information over a wide temperature range of 300–2,500 K. The gas phase thermal forward and reverse rate constants of the title reactions were calculated using the canonical variational transition state theory (CVT) and the microcanonical variational transition state theory (μ VT).
Methodology
Electronic structure calculations
The reactions of n-propyl radical and isopropyl radical with H atom are typical radical–radical reaction. It has been shown that DFT calculations or hybrid DFT calculations that mix in some Hartree–Fock exchange yield reasonable geometries, vibrational frequencies, and enthalpies of formation. While the configuration interaction (CI) method with the inclusion of triple excitations (QCISD(T) can provide quite reasonable energetics. Thus, the geometric parameters of all stationary points for the title reactions were computed using BHandHLYP approaches with the Dunning’s correlation consistent basis set including double-zeta, namely, cc-pVDZ. Here BHandHLYP denotes a combination of the hybrid Becke’s half-and-half (BHandH) method for nonlocal exchange and the Lee–Yang–Parr (LYP) nonlocal correlation functional. Another computational advantage of hybrid DFT method would allow the application of the direct dynamics method to the reactions involving large polyatomic molecules. The BHandHLYP/cc-pVDZ level of theory has been successfully applied to optimize the geometrical parameters and to calculate the harmonic vibrational frequencies of many species, particularly, only consisting of hydrogen and carbon elements. The MEPs were calculated using the intrinsic reaction coordinate (IRC) theory in mass-weighted Cartesian coordinates with a gradient step size of 0.01 (amu)1/2 bohr using BHandHLYP/cc-pVDZ level of theory. In particular, since no barrier was found at the UBHandHLYP/cc-pVDZ level of theory, we briefly discuss the method employed for calculating the MEP. Based on the physical nature of the title reaction that the distance between the forming H–H bond can be taken as distinguished reaction coordinate, we first fixed the forming H–H bond at large distance and optimize all other degrees of freedom of the reacting system. At this point, since all the forces were relaxed except for the fixed H–H bond due to the partial optimization, the combined force can be approximated as the force of the fixed bond. Thus, the direction of the combined force points to the direction of forming the products. We then start at this point to calculate the downhill IRC using the algorithm proprosed by Gonzalez and Schlegel. This is similar to the situation that one restarts an IRC calculation at an intermediate point of IRC. The IRC calculated using this method might not be, but possibly a good approximation to, the IRC path. Similar approximations were also adopted for radical–radical reactions without reaction barriers. Using this approximation, a maximum on the free energy curve at a given temperature generally serves as a pseudo-transition state that dominates the rate constants. At the points along the minimum energy paths, the force constants and Hessians were obtained to calculate the curvature of the reaction path and to calculate the generalized harmonic vibrational frequencies at the same level of theory. It is well-known that the accuracy of barrier height is very crucial for rate constant calculations. However, it has been observed that the DFT energies are not always sufficiently accurate for rate calculations. To obtain more accurate information on energy, QCISD(T)/cc-pVTZ method was employed to refine the energies of the points along the MEP optimized at the BHandHLYP/cc-pVDZ level of theory. The energy is denoted by QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ, where the double slash (X//Y) denotes geometry optimization at the level of theory Y and energy calculated at the level of theory X. Here QCISD(T)/cc-pVTZ is referred to as the Quadratic CI calculation including single and double substitutions with a triples contribution to the energy added using Dunning’s correlation consistent polarized valence triple-zeta basis set. For convenience, we denote the method as QCISD(T)//BHandHLYP. All the above calculations were performed using the Gaussian 98 program suite.
Rate constant calculations
Variational transition state theory is based on the idea that by varying the dividing surface along a reference path to minimize the rate constant, one can minimize the error due to “recrossing” trajectories. In the present study, the reference path is the MEP, which is defined as the steepest descent path from the saddle point to both the reactant and product directions in the mass-weighted Cartesian coordinate system.
For a canonical ensemble at a given temperature T, the CVT rate constant for a bimolecular reaction is given by
where
where kGT (T,s) is the generalized transition state theory rate constant at the dividing surface which intersects the MEP at s and is orthogonal to the MEP at the intersection point s. σ is the symmetry factor accounting for the possibility of more than one symmetry-related reaction path. In the case of the title reactions, we took the symmetry factor of 2 and 6 in the calculations of rate constants for the n-C3H7 + H and iso-C3H7 + H, respectively. β is (kB T)−1, where kB is the Boltzmann’s constant; h is the Planck’s constant. ΦR(T) is the reactant partition function (per unit volume for bimolecular reactions). VMEP(s) is the classical potential energy (also called the Born–Oppenheimer potential) along the MEP with its zero of energy at the reactants, and QGT(T, s) is the internal partition function of the generalized transition state at s with the local zero of energy at VMEP(s). Both ΦR(T) and QGT(T,s) partition functions are approximated as products of electronic, vibrational, and rotational partition functions. For the electronic partition function, the generalized transition state electronic excitation energies and degeneracies are assumed to be the same as at the transition state.
The μ VT is based on the idea that by minimizing the microcanonical rate constants k(E) along the MEP, one can minimize the error caused by the “recrossing” trajectories. Within the framework of μ VT, the rate constant at a fixed temperature T can be expressed as
where QR is the total reactant partition function, which is the product of electronic, rotational, and vibrational partition functions. The relative translational partition function was calculated classically and included in QR. However, the rotational and vibrational partition functions of the reactant were calculated quantum mechanically within the rigid rotor and harmonic oscillator approximations, respectively. Namely, the partition functions were calculated by directly sum over the quantum mechanical energy levels of rotational and vibrational motions. NGTS (E,s) is the sum of states of electronic, rotational, and vibrational motions at energy E of the generalized transition state located at s, where s is the reaction coordinate. NGTS (E, s) along the MEP was also calculated quantum mechanically using the rigid rotor and harmonic oscillator approximations. Furthermore, since no barrier was found on the MEP of the QCISD(T)//BHandHLYP level of theory, the tunneling effect for the title reactions was not considered in the present study.
The forward and reverse rate constants of the title reactions were calculated using two kinds of theory, namely, the CVT and the μ VT. The CVT and μ VT rate constants were calculated employing the online kinetics program VKLab.
Results and discussion
Stationary points
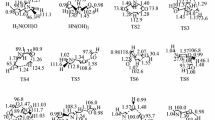
Pictorial diagram of optimized geometries of the stationary points are shown in Fig. 1 The properties of all the stationary points were previously computed at different levels of theory by many researchers. Table 1 lists the optimized geometric parameters of the reactants and products of the two reactions at the BHandHLYP level of theory with cc-pVDZ basis set along with the available experimental and theoretical data. For the stable points, it can be seen that both bond lengths and bond angles of propylene molecule predicted at the BHandHLYP level of theory are in good agreement with the experimental data, and geometric parameters of n-C3H7 and iso-C3H7 calculated using QCISD method are consistent with the theoretical data. Thus, it can be inferred that the BHandHLYP level of theory are suitable for predicting the geometries of stable species for the reaction. Due to the nature of radical–radical reaction, as is expected, no transition state was found at the BHandHLYP level of theory for the two reactions.

Pictorial diagram of optimized geometries of the stationary points.
Table 2 lists the harmonic vibrational frequencies and zero-point energies of reactant and products using BHandHLYP method with the basis set cc-pVDZ along with the available experimental data. From Table 2 one can find that the values of the calculated frequencies at the BHandHLYP level of theory are consistently large compared to the corresponding experimental values. However, the discrepancy between theoretical results and experimental data are generally within 7%. For example, the theoretically calculated harmonic vibrational frequencies (3,147, 3,116, and 3,108 cm−1) for n-C3H7 are all CH stretch, which are assigned to the available experimental values (3,100, 3,018, and 2,812 cm−1), respectively. The predicted harmonic vibrational frequencies (3,130, 3,128, 3,047, and 3,043 cm−1) for iso-C3H7 are CH stretch, which correspond to the available experimental values (3,052, 2,920, 2,850, and 2,830 cm−1), respectively. The predicted CH3 rock (1,186 cm−1) is assigned to the experimental value (1,165 cm−1). The predicted CCC bend for n-C3H7, iso-C3H7 and C3H6 are 340, 367, and 435 cm−1, respectively.
The two reactions energetics information including the reaction energies (E) and the reaction enthalpies ΔH°298 K at the BHandHLYP/cc-pVDZ and QCISD(T)//BHandHLYP levels of theory are listed in Table 3 along with the available experimental data. As can be seen from Table 3, the predicted reaction enthalpies (−63.76 and −59.72 kcal mol−1)(298 K) for the two reactions at the BHandHLYP/cc-pVDZ level of theory are much larger than the corresponding experimental values of −72.12±0.5 and −69.22±0.5 kcal mol−1, respectively. As is expected, the refined reaction enthalpies (−72.10 and −70.26 kcal mol−1) at the QCISD(T)/cc-pVTZ levels of theory are close to the corresponding experimental values for the two reactions. This implies that the reaction enthalpies can be effectively improved by the QCISD(T) method based on the BHandHLYP optimized geometries. For the two reactions, no barrier was found at the BHandHLYP/cc-pVDZ level of theory. When we refine the energies using the QCISD(T)/cc-pVTZ method, no barrier exists on the MEPs of the QCISD(T)//BHandHLYP level of theory either.
Reaction path properties
In this study, the minimum-energy paths of the two reactions were obtained using the intrinsic reaction coordinate (IRC) theory at the BHandHLYP/cc-pVDZ level of theory and the MEPs were further refined at the QCISD(T)/cc-pVTZ level of theory. Figure 2 presents the classical potential energy curves (VMEP) of the two reactions as a function of bond distance of the reactive moiety H–H at the QCISD(T)//BHandHLYP level of theory. To achieve an explicit comparison for the two reactions, a function of bond distance of the reactive moiety H–H of the corresponding VMEP is adopted instead of the reaction coordinate due to no transition state. Thus, the downhill algorithm was adopted to calculate the IRC at the UBHandHLYP/cc-pVDZ level of theory in contrast to the normal IRC calculation at the UQCISD/cc-pVDZ level of theory. As can be seen, the two VMEP and V Ga curves are strictly downhill in shape and very close to each other at above level of theory, namely, the transition states for the two reactions do not exist at the QCISD(T)//BHandHLYP level of theory, indicating that the tunneling effect is very small. Therefore, the tunneling effect was not taken into account for the two title reactions. This also implies that the positions of variational transition states for the two reactions will be close to each other at the corresponding temperature.

The classical potential energy (VMEP) and the ground state vibrationally adiabatic potential energy (V Ga ) curves of the reaction as a function of bond distance (Å) of the reactive moiety (H–H) for the two reactions at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory.
For barrierless reactions, the rate constant can be calculated by means of maximum of activation free energy ΔG(T,s) along the MEP on basis of canonical variational transition state theory. The free energy ΔG(T,s) vs temperature (T) and bond distance of the reactive moiety H–H at the QCISD(T)//BHandHLYP level of theory are plotted in Fig. 3 for the title reactions. It can be seen from Fig. 3 that the activation free energy for the reaction n-C3H7 + H (C3H6 + H2 are 22.23(s=0.98), −30.38(s=1.10), −50.37(s=1.18), and −69.98(s=1.18) at temperatures 300, 600, 900, and 1,500 K, respectively; while the activation free energy for the reaction iso-C3H7 + H (C3H6 + H2 are 21.87(s=0.99), −30.86(s=0.99), −50.85(s=1.07), and −70.48(s=1.07) at temperatures 300, 600, 900, and 1,500 K, respectively. It is obvious that the activation free energy varies with temperature and reaction coordinates and dominates the rate constant at different temperatures according to canonical variational transition state theory.

The free energy (G(T,s)) vs temperature (T) and bond distance of the reactive moiety H–H at the QCISD(T)//BHandHLYP level of theory: (a) reaction n-C3H7 + H (C3H6 + H2; (b): reaction iso-C3H7 + H (C3H6 + H2. Note that the arrow points to activation free energy at different temperature.
Rate constant calculations
The forward and reverse rate constants of the title reactions are calculated using the CVT and the μ VT employing the QCISD(T)//BHandHLYP potential energy information within the temperature range of 300–2,500 K. The calculated forward and reverse rate constants are shown in Figs. 4 and 5, respectively, together with available theoretical results. It can be seen from Fig. 4 that the two reaction forward CVT and μVT rate constants calculated from QCISD(T)//BHandHLYP potentials show positive temperature dependence in the temperature range 300–2,500 K, while the reported rate constants from Tsang are independent of temperature in the above temperature range. As is theoretically expected, the rate constants predicted from μVT are slightly smaller than those predicted from CVT in the calculated temperature ranges. Moreover, when temperature increases, the two reactions CVT and μVT rate constants predicted from the QCISD(T)//BHandHLYP method become consistent with each other, respectively. In the temperature range of 300–400 K, the forward rate constants predicted from CVT and μVT method are in excellent agreement with literature values from Tsang. From Fig. 5, the two reactions reverse rate constants predicted from CVT method are very close to those predicted from μVT method over the calculated temperature ranges. Table 4 lists the two reactions position of variational transition state calculated from QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory based on the CVT calculations. We can see that the variational transition state for the two reactions approaches to the product (R(H–H) decreases) when temperature increases. However, the variational effect of the two reactions is small at the QCISD(T)//BHandHLYP level of theory below 500 K. The positions of variational transition state for the two reactions are around 1.6 Å of R(H–H) over the temperature range of 300–500 K. This implies that we can use the geometry at this bond length to approximate the transition state and to calculate the rate constant for a wide temperature range without introducing large error, which will be very useful in predicting the rate constant of similar reactions based on the RC-TST theory. The fitted three-parameter Arrhenius expression of the calculated CVT rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory are kCVT (n-C3H7)=1.68×10−14 T0.84 e(319.5/T) cm3 molecule−1 s−1 and kCVT (iso-C3H7)=4.99×10−14 T0.90 e(159.5/T) cm3 molecule−1 s−1 for n-C3H7 + H and iso-C3H7 + H, respectively.

Comparison of the calculated forward rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory and the available experimental and theoretical data of the reaction vs 1,000/T for the two reactions.

Comparison of the calculated reverse rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory of the reaction vs 1,000/T for the two reactions.
Summary
We present a direct ab initio dynamics study of thermal rate constants of the two direct hydrogen abstraction reactions of hydrogen atom with n-propyl radical and isopropyl radical. BHandHLYP/cc-pVDZ method was employed to optimize the geometries of stationary points as well as the points on the MEP. The energies of all the points were further refined at the QCISD(T)/cc-pVTZ levels of theory. For the n-C3H7 + H and iso-C3H7 + H reactions, no one barrier was found at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory. The forward and reverse rate constants were evaluated with both CVT and μ VT in the temperature range of 300–2,500 K. The calculated results show that the rate constants have positive temperature dependence in the temperature range 300–2,500 K. The calculated forward rate constants at QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory are in good agreement with available literature data. The fitted three-parameter Arrhenius expression of the calculated CVT rate constants at the QCISD(T)/cc-pVTZ//BHandHLYP/cc-pVDZ level of theory are kCVT (n-C3H7)=1.68×10−14 T0.84 e(319.5/T) cm3 molecule−1 s−1 and kCVT (iso-C3H7)=4.99×10−14 T0.90 e(159.5/T) cm3 molecule−1 s−1 for n-C3H7 + H and iso-C3H7 + H, respectively. The positions of variational transition state for the two reactions are around 1.6 Å of R(H–H) over the temperature range of 300–500 K. We hope that this will be very useful in predicting the rate constant of similar reactions based on the RC-TST theory.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No: 20373007) and Foundation for basic research by Beijing Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Q.S., Zhang, Y. & Zhang, S. Direct ab initio dynamics studies of the hydrogen abstraction reactions of hydrogen atom with n-propyl radical and isopropyl radical. J Mol Model 11, 41–47 (2005). https://doi.org/10.1007/s00894-004-0218-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-004-0218-5