
Abstract
Molecular recognition between 4aS/R-galanthamine diastereoisomers (1: 4aS-galanthamine; 2: 4aR-galanthamine) and α-cyclodextrin (α-CD) were studied by use of docking and molecular dynamics (MD) simulation approaches. The binding energy of constructed 2···α-CD complexes is ~17 kcal mol−1 lower than that of 1···α-CD, implying a stronger binding ability of 2 with α-CD than that of 1. The theoretical modeling result is consistent with our previous CZE result, which demonstrated that α-CD is an efficient chiral additive for separating 1 and 2. The modeling result also indicates that both hydrophobic interaction and H-bond force may work as major factors for molecular recognition between the galanthamine diastereoisomers and α-CD.
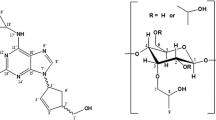
Figure Chemical structures of 4aS-galanthamine (left) and 4aR-galanthamine (right)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
4aS-Galanthamine, a tertiary alkaloid isolated from Amaryllidaceae, is a centrally acting, competitive, and reversible inhibitor of acetylcholinester (AChE), which enhances cognitive functions in Alzheimer’s patients. [1] Although it has been used in the past for the treatment of a variety of neurological disorders, it is currently attracting more attention as a possible agent in the treatment of Alzheimer disease (AD). This compound has excellent pharmacological and pharmacokinetic profiles and exhibits very low hepatotoxicity and side effects. [2, 3, 4] A number of total synthesis procedures are presently available and the chromatographic techniques for galanthamine detection, such as HPLC, HPLC-UV and HPLC-MS are well established. [5, 6, 7, 8, 9] However, little work has been reported on the chiral separation of 4aS/R-galanthamine diastereoisomers. With the fact that the inhibition of AChE is enantiomerselective, 4aR galanthamine should be strictly controlled for medicinal use. Our previous experiments have proved that α-CD is an efficient chiral additive for separating 4aS/R-galanthamine diastereoisomers using capillary zone electrophoresis (CZE). [10] The diastereoisomers show a ~1.4 fold difference in binding affinity for α-CD: K 1···α-CD=23.90 (mol l−1)−1, K 2···α-CD=33.98 (mol l−1)−1. As an aid in attempting to obtain structural insight into the possible recognition and binding mechanism between 4aS/R-galanthamine diastereoisomers with α-CD, molecular modeling procedures were performed here by separate docking runs of molecule 1 or 2 into α-CD. The located complexes with the highest affinity were then used as starting structures for molecular dynamics (MD) simulation in order to examine in more realistic detail how the diastereoisomers bind to α-CD. The binding energy of 1 or 2 with α-CD was calculated with Eq. (1)
where E complex is the total energy of the complex, E isomer is the energy of the global minimum conformation of 1 or 2, and E CD is the energy of the free α-CD. The chemical structures of compounds 1 and 2 are shown in Fig. 1. The four rings in the molecule are defined as ring I, II, III and IV, respectively.

Chemical structures of 4aS-galanthamine (left) and 4aR-galanthamine (right)
Experimental
All molecular modeling studies were performed on a Silicon Graphics O2 computer running MSI InsightII software. [11, 12] The basic modeling methodologies leading to the molecular dynamics (MD) trajectories and the energy-minimized complexes were performed using the CVFF [13] molecular mechanics force field implemented in the Discover_3 module of InsightII. Energy minimization was performed using the standard steepest descent and conjugated gradients minimization algorithms implemented in the program. For exhaustive conformational analysis of both galanthamine diastereoisomers, molecular dynamics simulation at 800 K was carried out. For molecular modeling of the interaction between 1 (or 2) with α-CD, molecular dynamics simulation at 300 K was performed and both galanthamine and α-CD were allowed to relax in these MD procedures.
Results and discussion
Molecular structures of 4aS/R-galanthamine and α-CD
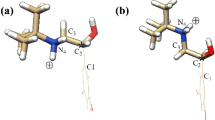
The published crystal structures of 4aS-galanthamine and its derivatives indicate that the four-ring system of galanthamine has a rigid conformation. [14] Therefore, the structure of 1 was obtained directly by extracting galanthamine from the crystal structure of complex of galanthamine with AChE. [15] An exhaustive conformational analysis of both 1 and 2 was also carried out by the molecular dynamics simulation method. The system was heated to 800 K for 5 ps, and MD trajectories for 10 ps were performed after that. A time step of 1 fs was used and the coordinates were saved at every 0.1 ps. Cluster analysis and minimization of the resulting conformers were performed for 1 and 2. The most stable conformations of 1 and 2 are shown in Fig. 2. The energy of the most stable conformation of 1 is 73.3 kcal mol−1 and that of 2 is 101.7 kcal mol−1. One intramolecular hydrogen bond is present in the most stable conformer of 1 between the hydroxy group and the O atom of the dihydrofuran ring (O6---O4=2.88 Å, O6–H6---O4=136.06°), which is similar to the correspondent crystal structures. The backbone structure of 1 obtained by the above MD analysis fits well with the published crystal structure, [15] the RMSD of all corresponding backbone atoms was 0.09 Å, indicating that the MD procedure used in this project is reasonable for the conformational analysis of galanthamine.

The most stable conformation: 4aS-galanthamine with E=73.3 kcal mol−1 (left), 4aR-galanthamine with E=101.7 kcal mol−1 (right)
The most stable conformations of 1 and 2 were superimposed using the InsightII superimpose module. All backbone atoms of rings II, III and IV were selected as comparison atoms. The calculated RMSD is 0.27 Å, which indicates that the geometry of rings II–III–IV of molecule 1 is similar to that of molecule 2, whereas the orientation of the cyclohexene ring (ring I) of 1 is obviously different from that of 2 due to the different chirality of the C4a atom. In molecule 1, the angle between mean planes of ring I (plane equation: 3.285x+6.230y−7.099z+8.957=0.0) and ring II (6.089x−5.168y−6.018z+4.725=0.0) is 72.3°, whereas in molecule 2, the plane angle between the mean planes of ring I (4.436x+2.741y−8.533z+64.79=0.0) and ring II (6.645x+0.2961y+7.467z+77.92=0.0) is 19.9°. Therefore, unlike molecule 1, where the ring I is nearly vertical to ring II, ring I in molecule 2 is nearly in the same plan with ring II and the whole molecule looks much flatter than molecule 1. This structural difference may be the key factor for molecular recognition between the 4aS/R-galanthamine diastereoisomers and α-CD.
The structure of α-CD was obtained from the Cambridge Structure Databank, its CSD entry is CHXAMH03. [16] Conformational analysis was also done on it by dynamics simulation at 300 K and the energy of the most stable conformation located is 167.2 kcal mol−1. The internal cavity of CD, which is hydrophobic, is a key structural feature of the molecule, providing the ability to complex and accept a variety of guest molecules. [17] The guests must satisfy the size criterion of fitting at least partially into the cyclodextrin cavity to form an inclusion complex. The inner diameter of α-CD is 5.7 Å, which may be suitable to accommodate a phenyl group, a cyclohexene ring or a tetrahydrozepine ring of galanthamine selectively. The selectivity of α-CD ring for rings of galanthamine is a crucial factor for separating galanthamine diastereoisomers.
Docking
The initial three-dimensional structures of the 1···α-CD and 2···α-CD complexes were generated manually by placing the galanthamine into the cavity of α-CD using computer graphics by means of the Insight II Docking module. The positions of galanthamine relative to α-CD were scanned by moving the galanthamine manually on the screen while InsightII calculates and displays energies. [18] The captured structure with the lowest energy was considered as the starting docking conformation for further molecular dynamics simulations. Both galanthamine and α-CD are held rigid during the interactive docking.
Molecular dynamics
In order to examine in more realistic detail how galanthamine diastereoisomers bind to α-CD, molecular dynamics (MD) simulations at a constant temperature of 300 K were performed on 1···α-CD and 2···α-CD complexes obtained by the above docking procedure. After the initial 5 ps heating period and temperature stabilization, MD trajectories were run for 100 ps. A time step of 1 fs was used and the coordinates were saved at every 1 ps. During the MD simulations all the 1, 2 and CD molecules were allowed to relax. Cluster analysis and minimization were performed on the resulting 1···α-CD and 2···α-CD conformers. Two typical conformers were found for both 1···α-CD and 2···α-CD. The molecular structures of these four typical conformers are shown in Figs. 3 and 4. The energy of the most stable conformer of 1···α-CD is 220.5 kcal mol−1, and that of the other conformer 221.4 kcal mol−1. The energy of the most stable conformer of 2···α-CD is 232.3 kcal mol−1, and that of the other conformer 239.0 kcal mol−1. The binding energy of 1···α-CD is about −20 kcal mol−1 and that of 2···α-CD about −37 kcal mol−1 according to Eq. (1). The calculated binding energy of 2···α-CD complex is up to 17 kcal mol−1 lower than that of the 1···α-CD complex. The result indicates that the binding ability of 2 with α-CD is stronger than that of 1, which is consistent with our CZE results. The different interactions of 1 with α-CD and 2 with α-CD resulting from the different geometry of 1 and 2 may form the basis for their chiral separation.

Typical conformations of 1···α-CD: the most stable conformation with E=220.5 kcal mol−1 (left), the other conformation with E=221.4 kcal mol−1 (right)

Typical conformations of 2···α-CD: the most stable conformation with E=232.3 kcal mol−1 (left), the other conformation with E=239.0 kcal mol−1 (right)
In the two typical conformers of the 1···α-CD complex, either the tetrahydroazeping ring (ring IV) or the cyclohexene ring (ring I) insert into α-CD’s hydrophobic cavity from the broad side of the cavity. There are two H-bonds found between the methoxy group of 1 and hydroxy group of α-CD in one of the typical conformers. Therefore, both hydrophobic interaction and H-bond force are contributing factors for the formation of the 1···α-CD complex.
In the most stable conformer of the 2···α-CD inclusion complex, the benzene ring (ring III) is deeply inserted from the broad side and oriented toward the narrow side of the α-CD’s hydrophobic cavity. In the other conformer, the hydrophobic cyclohexene ring (ring I) is inserted into the cavity of α-CD in a similar orientation. There is no H-bond found between 2 and α-CD. The hydrophobic interaction between guest and host is the main binding force for the formation of stable 2···α-CD complex.
Abbreviations
- Galanthamine:
-
4aS,6R,8aS-4a,5,9,10,11,12-Hexahydroxy-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-e,f]benzazepin-6-ol
References
Lyseng-Williamson KA, Plosker GL (2002) Pharmacoeconomics 20:919-942
Harvey AL (1995) Pharmacol Ther 68:113–128
Raskind MA, Peskind ER, Wessel T, Yuan W (2000) Neurology 54:2261–2268
Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C (2000) Neurology 54:2269–2276
Kuenburg B, Czollner L, Frohlic J, Jordis U (1999) Org Process Res Dev 3:425–431
Guillou C, Beunard JL, Gras E, Thal C (2001) Angew Chem Int Ed 40:4745–4746
Trost BM, Tang W (2002) Angew Chem Int Ed 41:2795–2797
Node M, Kodama S, Hamashima Y, Baba T, Hamamichi N, Nishide K (2001) Angew Chem Int Ed 40:3060–3061
Ingkaninan K, de Best CM, van der Heijden R, Hofte AJ, Karabatak B, Irth H, Tjaden UR, van der Greef J, Verpoorte (2000) J Chromatogr A 872:61–73
Yan LS (2003) PhD Thesis, Tsinghua University
Molecular Simulations Inc(MSI) (1997) InsightII 97, San Diego, Calif.
Molecular Simulations Inc(MSI) (1997) Discover_3 version 2.98, San Diego, Calif.
Dauber-Osguthorpe P, Roberts VA, Osguthorpe DJ, Wolff J, Genest M, Hagler AT (1988) Proteins Struct Funct Genet 4:31–47
Peeters OM, Blaton NM, De Ranter CJ (1997) Acta Crystallogr C 53:1284–1286
Bartolucci C, Perola E, Pilger C, Fels G, Lamba D (2001) Proteins Struct Funct Genet 42:182–191
Lindner K, Saenger W (1982) Acta Crystallogr B 38:203–205
Szejtli J (1998) Chem Rev 98:1743–1754
Molecular Simulations Inc(MSI) (1998) Affinity, San Diego, Calif.
Acknowledgements
This work was supported by the Chinese National Natural Science Foundation (No. 20132020 and No. 20175026), The Ministry of Science and Technology, The Chinese Ministry of Education and Tsinghua University, and The Tianjin Science Foundation (No. 023606511).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, M., Liu, X., Yan, L. et al. Molecular recognition between 4aS/R-galanthamine diastereoisomers and α-cyclodextrin. J Mol Model 9, 419–422 (2003). https://doi.org/10.1007/s00894-003-0162-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-003-0162-9